")

Rett syndrom
Rett syndrom är en sällsynt diagnos. Det beräknas finnas cirka 300 personer i Sverige med Rett syndrom. Handstereotypier, dyspraxi (svårigheter med viljemässiga rörelser) samt att barnen genomgår en period då de förlorar förvärvade förmågor i tidig ålder är typiskt för Rett syndrom. De allra flesta med diagnosen är flickor/kvinnor men det finns också ett litet antal pojkar/män.
Om Rett syndrom
Rett syndrom är oftast orsakat av mutationer i MECP2-genen på X-kromosomen. Diagnosen ställs utifrån kliniska kriterier men kan till cirka 95% bekräftas genetiskt. Över 300 olika mutationer i MECP2-genen är kända. I sällsynta fall kan andra genmutationer orsaka Rett syndrom och mutationer i MECP2-genen kan också förekomma vid andra diagnoser.
Möt individer och familjer
Att ta del av berättelser från andra familjer och individer som lever med Rett syndrom kan ofta vara värdefullt. Här har vi samlat ett urval av artiklar och filmer som vi hoppas att du ska ha glädje av. Kom ihåg att det finns en stor variation mellan individer och familjer samt att förutsättningarna är olika i olika länder. Varje individ har också sin unika personlighet. Tveka inte att ta kontakt med intresseorganisationer eller kontaktpersoner.
Inez och Polly har ovanliga diagnosen Retts syndrom – åker på läger i Trosa kommun varje sommar
I detta reportage från SVT Nyheter får vi följa Inez Carracedo och Polly Slotwinska som båda har Retts syndrom. De senaste åren har de deltagit i ett läger i Trosa kommun som anordnas för familjer med barn som har diagnosen. Notera att filmklippet i artikeln ej är tillgängligt längre.
Living with Rett Syndrome
Living with Rett Syndrome från Special books är filmer på engelska där vi får möta Brynn, Liliana med flera som lever med Rett Syndrom i USA.
Vi syns – Möt Cecilia
Vi får träffa Cecilia som bor i Sollentuna utanför Stockholm. Cecilia har Retts syndrom. Hon är en envis liten tjej som tycker om när det händer saker, att busa och bada. I den här filmen får vi följa Cecilia under en dag på förskolan.
Living with Rett Syndrome
Living with Rett Syndrome från Special books är filmer på engelska där vi får möta Brynn, Liliana med flera som lever med Rett Syndrom i USA.
Artikel – Ett barn med Retts syndrom behöver hjälp med allt
Läs artikeln om Vera som bor i Finland med sin familj. Mamma Anna berättar bland annat om betydelsen av stöd från andra familjer och engagemanget i anhörigföreningar.
Artikel – Tildas aktiva sommar
Tilda Alm har nyss fyllt 13 år. Hon tycker om när det händer saker och älskar att dansa, rida, umgås med kompisar och gå på konsert. De som följer hennes Instagramkonto @tildisbus får hänga med i svängarna, den här sommaren har det varit fullt upp! Att Tilda lever med Rett syndrom är inget hinder.
RSIS är en förening som drivs av anhöriga till personer med Rett syndrom. Vi arbetar för att bidra med information och kunskap om Rett syndrom, stödja föräldrar och anhöriga och vara ett nätverk. Vi hittar också på mycket roligt tillsammans!
![]()
International Rett Syndrome Foundation i USA är den största organisationen för Rett syndrom. De samlar in pengar till forskning, stödjer familjer och jobbar med opinionsbildning.
1. Vad har du för anknytning till Rett syndrom?
– Jag är mamma till en vuxen dotter med Rett syndrom.
2. Kan du berätta lite om din dotter?
– Lisa är 36 år. Hon lever ett aktivt liv – hon är med i en kör, hon bowlar, rider och träffar kompisar regelbundet. Båda Lisas syskon har tre barn var och för dem och oss alla är Lisa en självklar del i vår stora familj. Syskonbarnen tycker om att spela spel med Lisa på hennes ögonstyrda dator och de räknar alltid med henne när vi ska göra något med familjen. Lisa tycker om sol och värme och att resa. Vi har rest mycket tillsammans och inte låtit Lisas svårigheter stoppa oss. Vi har varit till Florida flera gånger och Lisa har badat med delfiner och hängt med på olika äventyr.
3. Hur gick det till när din dotter fick diagnosen?
– Jag började ana att något inte stämde redan när Lisa var tre månader gammal. Hon orkade inte lyfta sitt huvud, och jämfört med sin storasyster utvecklades hon långsamt. På BVC blev jag inte tagen på allvar – de sa att hon bara var lite sen. Vid ett års ålder påpekade jag att hon saknade fallreflexer och då började utredningarna. Det dröjde ytterligare några månader tills vi fick komma till habiliteringen där vi träffade en läkare som misstänkte Rett syndrom. Vi fick därefter en remiss till Bengt Hagberg i Göteborg, kanske den läkare i världen som då visste mest om Rett syndrom. Han sa direkt att det fanns en stark misstanke att Lisa hade Rett syndrom.
4. Hur var första tiden efter att ni fått diagnosen?
– Vi hade svårt att ta in beskedet om diagnosen. Från att BVC sagt att hon bara var lite sen till att få veta att hon troligen skulle få epilepsi, skolios och en hel rad andra utmaningar var svårt att ta in. Vi var inte redo och kände oss inte sedda som föräldrar. Efter en tid insåg vi att det ändå var Rett syndrom som Lisa hade.
Jag började engagera mig i Rett syndrom och var bland annat på världskongressen i Antwerpen 1993. Då kom jag i kontakt med Bengt Hagberg igen och vi inledde en fin vänskap och ett gott samarbete tillsammans med Rett-sektionen som mynnade ut i en världskongress i Göteborg 1996. En kongress som vi inte hade kunnat göra utan honom och hans kontakter. I samband med världskongressen 1996 bildades RSIS som tidigare varit en sektion inom föreningen Autism.
5. Finns det något ni har saknat sedan ni fick diagnosen?
– I början saknade vi mycket, men det har hänt otroligt mycket sen dess – särskilt kring kommunikation och förståelsen för vad personer med Rett syndrom faktiskt kan. Idag får tjejer med Rett syndrom tidigt sin ögonstyrda dator och det gör en stor skillnad att kunna kommunicera med sin omgivning. Jag är glad att hon har en diagnos som det forskas mycket kring. Nu finns det medicinering framtagen i USA och fler mediciner liksom genterapi är på gång.
6. Hur ser vardagslivet ut idag och vad hoppas du på för framtiden?
– Vardagslivet rullar på bra, mycket tack vare att Lisa har haft samma fantastiska assistenter i många år. En har varit hos oss i över 30 år! Lisa har en stor trygghet i sina assistenter och lever tack vare dem sitt eget liv, vid sidan av vårt gemensamma familjeliv. Som jag berättade tidigare så har hon ett bra och aktivt liv. En sak som hon verkligen gillar är projektet ”Lyssna till mitt öga” där Lisa träffar andra personer med Rett syndrom och spelar musik tillsammans. De har olika teman, förra gången var det schlager, tidigare har de haft bl a klassiskt och rap som tema. De använder en app som heter Eyeharp som jag gärna rekommenderar.
7. Varför har du valt att ställa upp som kontaktperson?
– Jag har varit ordförande i RSIS i många år och har lång erfarenhet. Det föll sig naturligt att bli kontaktperson. Jag finns här för dem som behöver prata. Jag kan också slussa vidare till andra med specifik erfarenhet, till exempel om man har ett yngre barn och vill komma i kontakt med någon i samma situation. Det känns meningsfullt att kunna bidra. Och jag tycker att det är viktigt att vi i styrelsen representerar fler perspektiv än bara småbarnsåren.


Hälsa och utveckling
Alla människor har rätt till hälsa och utveckling genom hela livet. För personer som lever med Rett syndrom kan det innebära att det krävs både stöd för delaktighet samt behandling av de symtom som kommer med diagnosen. Det är viktigt att ha i åtanke att insatser behöver utformas utifrån kunskap om diagnosen, liksom varje individs personlighet, specifika behov och möjligheter.
Stöd för delaktighet
Stimulerande och utvecklande aktiviteter är viktiga förutsättningar för en god livskvalitet. Förhållningssätt och bemötande från människor i omgivningen samt medicinska åtgärder är också betydelsefulla för att upprätthålla fysisk och psykisk hälsa.
Insatser behöver utformas utifrån kunskap om diagnosen men även utifrån varje persons specifika förmågor och behov. Alla människor kan utvecklas inom olika områden oavsett ålder. En vardag med meningsfulla aktiviteter, rutiner och en balans mellan återhämtning och utmaningar behövs genom hela livet.
Forskning om aktiviteter och Rett syndrom
Flera studier har undersökt vad personer med Rett syndrom tycker om att göra. En studie visade att de gillade att bada, lyssna på musik och vara utomhus. De tyckte mindre om saker som tandborstning, hygien och viss motorisk träning. Föräldrar och personal gillade oftast samma aktiviteter som personerna med Rett syndrom.
En annan studie följde tio personer under en vecka. Den visade att mycket tid gick åt till hygien, mat, vila, vård och transport. Det fanns lite tid för fysisk aktivitet, skolarbete och socialt umgänge. När det gäller vilka personer individer med Rett syndrom tillbringade mest tid med, var det oftast personal, därefter familjen och i minst utsträckning vänner.
En dansk studie som undersökte delaktighet i olika familjeaktiviteter visade att personer med Rett syndrom var mest delaktiga i lugna aktiviteter inomhus. Utomhusaktiviteter var ovanliga, men när de hände var de mycket uppskattade. Vardagsrutiner var vanliga men inte lika engagerande.
Sammanfattningsvis visar forskning att personer med Rett syndrom mår bra av att vara delaktiga i såväl vardagsrutiner som stimulerande aktiviteter. Det är viktigt att anpassa aktiviteter så att de passar individen och görs mer engagerande. Exempel på anpassningar är att använda flera sinnen och att hjälpmedel, så som kommunikationsstöd, finns tillgängliga under aktiviteter
Att vara delaktig i vardagsaktiviteter och rutiner
Anpassa dagliga aktiviteter och rutiner så att personen med Rett syndrom kan vara delaktig och påverka på sitt sätt. Fundera på hur aktiviteterna kan utformas för att personen ska kunna vara så självständig som möjligt. Tänk också igenom vilka moment i aktiviteten personen själv kan vara aktiv i – på egen hand eller med stöd. Det kan till exempel handla om att:
- personen själv väljer vilka kläder denne vill ha på sig
- hjälpa till att ta på sig kläderna
- använda alternativa styrsätt (kontakter) för att delta i hushållssysslor så som matlagning.
- hjälpa till att bära/köra tvättkorg till tvättstugan
Fundera också över om personen med Rett syndrom till exempel kan uttrycka vad hen tycker om det ni gör; om det är roligt eller tråkigt, göra mer eller sluta.
Det är bra att tänka över om det är några hjälpmedel som behöver finnas med under aktiviteten. Till exempel kommunikationsstöd, tidshjälpmedel eller schema över det som ska göras.
Fundera också över om det finns en balans mellan aktiva och mer lugna, återhämtande moment, så att dagens aktiviteter både ger utmaningar och återhämtning.
Samspelsstöd i aktiviteter
Att samspela och göra aktiviteter tillsammans är roligt! Vissa personer med Rett syndrom kan behöva stöd för att samspela och delta, till exempel genom att vi i omgivningen är inlyssnande och uppmärksamma på vad personen är intresserad av för tillfället, och utgår ifrån det. Följande strategier kan vara gynnsamma att tänka på i samband med aktiviteter med personer med Rett syndrom:
- Repetition och tydliga pauser i till exempel aktiviteter kan locka till svar och initiativ från personen med Rett syndrom. Många personer med Rett syndrom kan ha en svarslatens som gör att det kan ta olika lång tid innan ett svar eller en reaktion kommer.
- Pauser kan också skapa spänning och förväntan, vilket kan motivera och locka personen att själv vara mer aktiv. Detta är i sin tur en viktig förutsättning för att utveckla såväl kommunikativa, motoriska som kognitiva förmågor. Ett exempel kan vara att göra paus när ni gungar, kastar boll eller sätter på nästa favoritlåt. Vänta in att personen med Rett syndrom visar –på det sätt hen kan – att hen vill göra mer.
- Att vi i den sociala omgivningen är med och deltar i aktiviteter och bidrar med lust och energi. Många personer med Rett syndrom tappar ibland i vakenhet och fokus och kan då behöva hjälp att återengagera sig i aktiviteten. Naturligtvis behöver det finnas en balans mellan aktivitet och återhämtning.
Det finns flera tips på samspel att använda i olika aktiviteter på länkarna nedan. Pröva gärna strategierna när ni gungar, hoppar på studsmatta eller kanske spelar musik tillsammans.
Förslag på aktiviteter
Att ha något meningsfullt och roligt att göra är viktigt för att må bra. Vad vi tycker är roligt är väldigt olika och nedan följer förslag på aktiviteter.
Aktiviteter i naturen
Att vara ute i naturen tillsammans är bra både för kropp och själ. Det finns många aktiviteter ni kan göra tillsammans utomhus. Förutom mer säsongsbetonade aktiviteter såsom bär- och svampplockning, vinter- och sommarsport, så kommer här lite fler tips på vad ni kan göra under er utomhusvistelse:
- Säsongsbingo: En rolig aktivitet är att leta efter olika tecken i naturen när en säsong ger plats åt en ny. Vi på Nationellt Center har tagit fram säsongsbingo som ni kan pröva på nästa promenad.
Vår säsongsbingo - Fotografera och återberätta – En annan aktivitet är att fotografera och sedan lägga in det i ett bildspel för att samtala och återberätta om det ni har varit med om.
- Att göra en hinderbana i naturen är också ett roligt sätt för en lite mer aktiv och rolig utomhusvistelse. På Eldorado resurscenters hemsida hittar ni mer inspiration kring vad ni kan hitta på.
Läs mer - Ett annat tips är att följa en orienteringskarta eller snitslad bana.
- Spela frisbeegolf är en annan rolig aktivitet. Gå och kasta eller lägg frisbeen i nära och synliga mål.
Rörelse- och dansaktiviteter
Att delta i rörelse- och dansaktiviteter är roligt och har många positiva effekter. Vi kan både stärka och utveckla våra muskler och bli uppiggade och avslappnade. När vi dansar tillsammans och får beröring så frigörs också vårt lugn-och–ro-hormon, oxytocin.
Det finns många gymnastik- och friskvårdsföretag som anordnar träning och dans för personer med olika funktionsnedsättningar:
För mer tips om dans för personer med olika funktionsnedsättningar hänvisas till: Klangfärg i Västra Götalandsregionen som samordnar tips och idéer kring kulturaktiviteter med bland annat dans.
För personer med omfattande motoriska funktionsnedsättningar kan materialet och metoden “Liggande dans” vara intressant.
Aktiviteter med färg och form
I aktiviteter med färg och form får vi utforska och uppleva olika material och sinnesförnimmelser, vilket kan ge glädje i stunden och ökad erfarenhet av stimulans till utveckling. I materialet ”Art så klart” som är gjort av en arbets- och bildterapeut, ges 25 tips på hur du kan assistera i bild och färg och form aktiviteter av väldigt olika slag – här finns något för alla!
Fler tips på färg- och formaktiviteter att göra tillsammans ges på Klangfärgs webbplats.
Övriga aktivitetstips
Fritidsinfo fritidsaktiviteter för dig med funktionsnedsättning – Webbplats där det går att hitta olika typer av aktiviteter för personer med funktionsnedsättning i hela landet.
Kognitivt stöd underlättar att förstå, minnas, planera och hantera tid. Stödet kan vara i form av andra människor, strategier, anpassningar i miljön eller olika hjälpmedel. Vid Rett syndrom kan exempelvis benägenhet till oro göra att kognitivt stöd blir extra viktigt för att göra vardagen mer hanterbar och begriplig. Nästan allt stöd behöver på ena eller andra sättet individanpassas.
Vi behöver alla kognitivt stöd i någon form. Det kan handla om olika typer av stöd för tanke och minne, såsom schema eller kalender för vad vi ska göra, eller någon typ av tidshjälpmedel. Kognitivt stöd kan se ut på många olika sätt, kompensera för olika svårigheter och därigenom underlätta vardagen.
Vad är kognition?
Kognition är ett sammanfattande ord för människans förmåga att lära, tänka och bearbeta information. Kognition innehåller många olika delar, som alla är viktiga för att vardagen ska fungera.
Funktioner som är kopplade till kognition är till exempel:
En intellektuell funktionsnedsättning påverkar hur personen förstår sin tillvaro, det vill säga verklighetsuppfattningen, rumsuppfattning, tidsuppfattning, kvalitets- och kvantitetsuppfattning samt orsaksuppfattning. För personer med en mer omfattande intellektuell funktionsnedsättning behövs ofta konkret information för att personen ska kunna agera självständigt. Det kan också vara stödjande om händelser sker på samma sätt och stegvis så att personen ges förutsättningar att förstå och vara delaktig i det som sker.
När det gäller rumsuppfattning kan det till exempel handla om att det finns tydliga riktmärken i miljön, bilder eller föremål utanför olika dörrar i en korridor eller att ni går samma väg varje gång ni ska till en specifik plats.
När det gäller tidsuppfattning behöver många personer med intellektuell funktionsnedsättning få tid presenterad för sig på ett mer konkret och visuellt sätt för att förstå när något ska inträffa, och i vilken ordning saker och ting sker. Tidsuppfattningen är inte statisk utan utvecklas hela tiden. Utveckling underlättas genom redskap för att förstå tid, genom rutiner i vardagen och genom att prata och resonera om tid.
När det gäller orsaksuppfattning, det vill säga att förstå orsak och verkan, är det enklast när en egen handling har direkt effekt, såsom att trycka på en touchkontakt som spelar upp musik. Om en person har en intellektuell funktionsnedsättning kan det däremot vara svårare att förstå orsak och verkan som inte följer direkt på varandra.
Det är svårt att bedöma exakt hur varje individ med Rett syndrom förstår sin tillvaro och den kognitiva förmågan hos olika personer med diagnosen varierar mycket. Studier där ögonstyrda datorer använts har visat att många personer med Rett syndrom kan förstå både visuell information och ord bättre än vad som visats i tidigare studier. Oavsett kognitiv förmåga kan kognitivt stöd ha stor betydelse för att ge personen trygghet och mer kontroll i sin vardag.
Varför är kognitivt stöd viktigt?
“Kognitivt stöd gör det osynliga synligt”
Med kognitivt stöd kan personer med Rett syndrom få ökad självständighet och kontroll genom att det synliggör och underlättar förståelse, minne och planering på olika sätt. Personen behöver därför inte ”hålla allt i huvudet” och avlastas på så sätt. Över tid kan kognitivt stöd som åskådliggör tid bidra till att tidsuppfattningen utvecklas.
En kognitivt tillgänglig miljö ger svar på viktiga frågor såsom:
Sammantaget kan kognitivt stöd medföra att personen får mer energi över till annat.
Kognitivt stöd
Kognitivt stöd kan vara:
Det kognitiva stödet behöver anpassas efter individens förmågor, behov och beroende på situation. För att stödet ska upplevas meningsfullt för individen är det viktigt att tänka utifrån dennes perspektiv. Vad tycker personen är viktigt att veta? Det är också viktigt att inte bara använda kognitivt stöd för att berätta om sådant omgivningen vill att personen gör utan också koppla det till aktiviteter eller företeelser som personen tycker om. Om möjligt är det bra om personen själv är med och bestämmer innehåll och utformning av de redskap som används.
Det är viktigt att introducera och stötta personen att använda sina kognitiva stöd, exempelvis titta gemensamt på schemat vad som ska göras inför en ny aktivitet.
Arbetsterapeut eller specialpedagog kan hjälpa till att utforma och introducera stödet.
Andra människor
För många med Rett syndrom utgör andra människor en stor del av det kognitiva stödet. Det är vanligt att personer i omgivningen är de som håller reda på tider och vad som ska hända under dagen och veckan. Mänskligt kognitivt stöd har den stora fördelen att det går att anpassa efter individen och situationen som uppkommer. Att hålla koll på trafiken för att avgöra när det är säkert att gå över gatan är ett exempel på stöd som troligtvis bäst sker med hjälp av andra personer. Andra personer håller också ofta reda på, och berättar, vad som ska hända. De instruerar, förklarar och visar på ett sätt de av erfarenhet vet fungerar bra, de använder exempelvis ett enkelt språk utan liknelser eller långa, krångliga meningar.
Det är viktigt att ha i åtanke att kognitivt stöd via andra människor riskerar att bli personbundet och förändras eller försvinna om exempelvis personal byts ut. När andra människor står för det kognitiva stödet, innebär det också att personen inte kan komma åt informationen på egen hand. I appen RättVisat går det att samla information kring en person digitalt. Ett annat förslag för nätverk med flera stödpersoner är aktivitetskort och aktivitetsdagbok. Dagboken ger en överblick över vilka aktiviteter personen gjort under veckan och aktivitetskortet visar vilken aktivitet kortet gäller, varför aktiviteten ska genomföras, vad som behövs och annat att tänka på. På hemsidan för aktivitet i projektet ”Vi-är-med” finns aktivitetskort och aktivitetsdagbok att fylla i samt exempel.
Miljön
Miljön kan anpassas för att ge kognitivt stöd. Att tydligt märka upp var personens kläder ska hänga eller personens plats kan svara på den viktiga frågan “Var ska jag vara? Bilder, föremål samt färger är bra att använda för att märka upp platser. Om personen har svårt att vänta kan en tydlig plats där personen kan sitta och vänta, till exempel på att bussen ska komma eller på att få gå in och göra något roligt som att bada, underlätta. Det kan också vara bra att se till att det finns något att göra där, exempelvis kan böcker eller några leksaker underlätta väntan.
Bildscheman över dagen (se nedan under hjälpmedel) som finns uppsatta på strategiska ställen så att personen enkelt kan gå och titta vad som ska hända är ett kognitivt stöd som många känner till.
Många personer med Rett syndrom har stort behov av regelbundna återhämtande aktiviteter, exempelvis på grund av hyperaktivitet eller avvikande perception. Att ha särskilda platser för återhämtande aktiviteter kan underlätta för personen att lätt komma ner i varv. På så sätt kan personer som har gångförmåga själva visa att en vila behövs genom att ta sig till dessa platser. En del personer kan tröttas av många intryck och då kan det vara värt att kartlägga miljöer där personer ofta vistas utifrån detta perspektiv för att se om de går att anpassa.
På Habilitering och Hälsa finns filmer som visar hur olika miljöer kan anpassas med kognitivt och kommunikativt stöd. Här ges också många exempel på hjälpmedel.
Hjälpmedel
Det finns många hjälpmedel för kognitivt stöd. En hel del går att köpa i allmän handel och en del kan förskrivas eller köpa från särskilda hjälpmedelsföretag. Reglerna för förskrivning varierar mellan olika regioner. För att få hjälpmedel förskrivna behöver du kontakta en arbetsterapeut. På bildstod.se går det att skapa ett konto och själv skapa bildstöd av olika slag. Där går också att söka bland andras bildstöd för att få inspiration eller ha som mall och anpassa.
Här är en film om visuellt stöd från Habilitering och Hälsa. Informationen är relevant även om filmen handlar om visuellt stöd vid autism.
Scheman och kalendrar kan utformas på olika sätt beroende på hur långt personen kan överblicka, vad som är viktigt för personen att veta etcetera.
I Sverige används följande färger för att representera dagarna: måndag – grön, tisdag – blå, onsdag – vit, torsdag – brun, fredag – gul, lördag – rosa och söndag – röd. Dagsscheman ger en överblick över dagen och kan hjälpa till att utveckla tidsuppfattning. De kan också hjälpa till att förbereda när rutiner bryts. På habilitering och hälsa finns filmer som visar hur olika miljöer kan anpassas med kognitivt och kommunikativt stöd.
För många är det viktigt att veta vem som ska hämta, vem som ska jobba om personen har assistenter eller boendepersonal.
Ett alternativ för personer som är hjälpta av talad information är att koppla en digital kalender till en röstassistent. Då kan personen själv med hjälp av ett talande kommunikationshjälpmedel fråga röstassistenten om vad som händer under dagen eller om annan information som kan finnas i en kalender.
Här kan du läsa om hur Anna, Agnes och Matilda som har Rett syndrom använder sig av sina smarta-hem-funktioner.
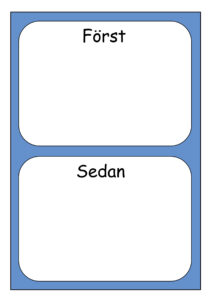
Ett först-sedan-stöd kan underlätta att göra sådant som är svårt eller tråkigt. I rutan för först sätts en bild på exempelvis ”borsta tänderna” och i rutan för sedan sätts en bild för något personen tycker om, exempelvis ”sitta i saccosäck med hörlurar”.
Val: En del personer med Rett syndrom går omkring till synes planlöst. Det kan fylla ett behov hos personen men ibland kan det också bero på att personen inte själv kan komma på något att göra. Det kan då underlätta att lägga in olika förslag på aktiviteter som personen kan välja bland i dagsschemat.
Aktivitetsschema bryter ner en aktivitet eller uppgift i mindre steg, exempelvis morgon- och kvällsrutin, att ta medicin eller gå på café.
Tidshjälpmedel: Att ha en uppfattning om tid är svårt för många med Rett syndrom. Här är en film om tidshjälpmedel från Habilitering och Hälsa. Informationen om att lära sig tid och tidshjälpmedel är relevant även om filmen handlar om autism.
Nedan finns länkar till firmor som säljer hjälpmedel för kognitivt stöd:
Bildstöd i vårdsituationer
Tips på mer information:
Berit Larsson, leg. arbetsterapeut och handledare, Eldorado resurscenter har granskat texten.
Kommunikation är viktigt för delaktighet. Omgivningens förväntningar på att personen med Rett syndrom kan lära sig nya sätt att kommunicera på har stor betydelse. Erfarenhet av världen, samspel, kommunikationshjälpmedel såsom ögonstyrda datorer och att få påverka sin tillvaro är också viktigt för utveckling av språk och kommunikation.
Om kommunikation och språk
Kommunikation innebär att två personer på olika sätt påverkar varandra i samspel med eller utan ord. Språket är de ord och begrepp vi kan, hur vi sätter ihop dem till meningar eller hela berättelser och hur vi använder vårt språk beroende på situation eller vem vi pratar med. I språk ingår också vilket sätt vi uttrycker orden på, exempelvis via tal, tecken eller bildsymboler. Utvecklingen av kommunikation och språk börjar så snart vi föds och utvecklas sedan hela livet. Flera viktiga steg i utvecklingen sker redan innan vi börjar använda tal för att kommunicera. Vanligtvis sker kommunikationsutveckling genom att personer i omgivningen tolkar alla barnets signaler till att barnet förstår att det kan påverka andra att göra saker. Så småningom lär sig barnet att använda språk för att kommunicera.
De olika stegen i den tidiga kommunikationsutvecklingen beskrivs ofta utifrån hur utvecklingen vanligtvis går till och att utvecklingen sker stegvis. För personer med Rett syndrom ser utvecklingen annorlunda ut. De befinner sig troligtvis på flera utvecklingssteg samtidigt och stödet från omgivningen är extra viktigt för utvecklingen. Att känna till de olika stegen och ungefär på vilket eller vilka steg en person verkar befinna sig på är till god hjälp för att stötta kommunikation och språk. Stegen beskrivs på lite olika sätt, men en vanlig beskrivning är att personen:
- Kommunicerar via spontana handlingar. Det är reaktioner på sådant som händer inuti och utanför kroppen som inte är viljestyrda eller målmedvetna. Om omgivningen svarar, kanske gör igen när personen verkar gilla något eller exempelvis ändrar sitt tonläge när personen inte verkar må bra, kan personen ges möjlighet att kommunicera behov eller sinnesstämningar.
- Använder medvetna handlingar – agerar viljemässigt och målinriktat för att påverka saker i omgivningen.
- Riktar sin kommunikation för att få någon annan att göra något särskilt.
- Använder bildsymboler, tecken eller talade ord för att kommunicera.
Kommunikativ utveckling
De första viktiga stegen i kommunikativ utveckling.
Kommunikation, språk och tal vid Rett syndrom
Personer med Rett syndrom behöver mycket stöd med sin kommunikation. Ett litet antal personer med Rett syndrom använder tal och färre personer använder tal i högre ålder. Cirka 5% kombinerar flera talade ord. Hos en del personer som talar kan det enligt vår erfarenhet förekomma att talet används sällan. Personen kan också säga ett ord vid ett enstaka tillfälle och sedan inte upprepa detta. De allra flesta med diagnosen förstår mer än vad de kan uttrycka. Det är därför viktigt att använda både kognitivt och kommunikativt stöd såsom bildscheman och kommunikationskartor för bästa möjliga förutsättningar till delaktighet och utveckling. Ögonstyrda datorer är ofta det mest tillgängliga sättet för att styra omgivningen och att uttrycka språk.
Riktlinjer för kommunikation vid Rett syndrom publicerades 2020 baserade på vetenskapliga studier och erfarenheter från föräldrar och professionella. Titta gärna också på vår film om kommunikation vid Rett syndrom.
Kommunikation påverkas av individens intresse för socialt samspel, motorik, kognition, allmänt hälsotillstånd, personlighet etc. Där är den individuella variationen stor och påverkas till viss del av omgivningen. I riktlinjerna understryks vikten av att introducera och använda kommunikationshjälpmedel. Det rekommenderas också att en bedömning görs utifrån varje individ. Hänsyn bör också tas till resurser och behov hos personerna i omgivningen för att de ska kunna ge bästa möjliga stöd. Ta kontakt med logoped på habiliteringen för att få stöd och samarbeta med förskola/skola eller daglig verksamhet.
Skapa förutsättningar för kommunikation
Att skapa förutsättningar för kommunikation handlar i mångt och mycket om att ge personen “något att prata om och någon att prata med”. Att se till att det händer saker som väcker intresse och engagemang är en bra start. Rutiner, välkänd miljö och välkända aktiviteter är viktigt och ger trygghet. När personer vet vad som förväntas kan de ofta delta på bästa möjliga sätt. Å andra sidan kan en alltför lugn och förutsägbar tillvaro skapa passivitet. Om allting utförs på rutin eller i en viss ordning ges inte utrymme för att påverka. Utforska vad som väcker intresse och hjälp till att vidga världen på olika sätt samt ge möjlighet att påverka inom mer eller mindre välkända ramar.
Se till att det finns saker i miljön att titta på, gå fram till, känna på. Om blicken fångas eller om personen går fram till något – tolka detta som ett initiativ till kommunikation. Benämn det som väcker intresse, titta tillsammans. Bilder och föremål som stöd i samtal underlättar gemensamt fokus och gör det lättare att prata om samma sak.
För alla individer är det viktigt att kunna påverka det som sker och att kommunicera sina önskemål, tankar och känslor till någon som är intresserad och nyfiken. Var engagerad, lyhörd, närvarande och fokuserad. Gör pauser och invänta svar i samspelet. Detta benämns ofta som att vara responsiv. Eftersom många med Rett syndrom behöver mycket tid för att kommunicera är det viktigt att ge tid att ta kommunikativa initiativ och att svara.
I kontakt med nya människor behöver personer med Rett syndrom hjälp med att föra en dialog så att de inte utesluts ur samtalen.
Här finns mer information om att vara responsiv :
Att påverka
Erbjud många tillfällen att påverka. Personer som inte använder så tydliga kommunikativa signaler kan ges möjlighet att påverka med sin blick, exempelvis i måltidssituationen genom att du väntar in att personen tittar på dig eller tallriken för att visa att det är dags för nästa tugga eller att personen tittar på dig eller boken för att visa att du ska läsa vidare eller berätta om en bild.
Alternativa och kompletterande kommunikationssätt
Alternativ och kompletterande kommunikation (AKK) används i stället för, eller som komplement till tal. En del AKK-sätt involverar bara kroppen och en del involverar även något slags redskap, kommunikationshjälpmedel. Många personer med Rett syndrom kommunicerar mycket med kroppskommunikation: fysiska reaktioner, rörelser, gester, mimik, ljud men kommunikationshjälpmedel av olika slag är nödvändiga för att kunna samtala om sådant som inte finns här och nu.
Bilder
Bilder kan användas på olika sätt – som kognitivt stöd för att förklara och underlätta förståelsen (till exempel i bildscheman), som fokus för samtal (exempelvis illustrationer i böcker, foton eller bilder i spelappar), samt som ett sätt att uttrycka sig. För att en person ska kunna uttrycka allt den vill krävs dock ett större ordförråd med bilder som språk. Bilder som språk är enkelt uttryckt bilder där det finns en bild för varje ord (eller fras).
Stora ordförråd
Stora ordförråd samlar många bildsymboler och/eller skrivna ord så att ord för olika situationer, ämnen och samtal med olika personer i omgivningen finns tillgängliga. Målet är att personen ska kunna kommunicera om vad som helst med vem som helst. Det är att rekommendera att använda något av de färdiga stora ordförråd som finns snarare än att göra ett eget. Core first, TD Express, PODD och Voco Chat är exempel på några av de stora ordförråd som finns på svenska. På DARTs hemsida finns bl.a. information om stora ordförråd.
Talande hjälpmedel
Talande hjälpmedel läser upp meddelanden som någon spelat in eller med hjälp av syntetiskt tal. De kan vara mycket enkla och uttrycka enstaka meddelanden eller vara avancerade och innehålla stora ordförråd. Talande hjälpmedel kan kombineras med bilder eller text och kan styras på olika sätt. Vid Rett syndrom är ögonstyrda datorer ofta det hjälpmedel som ger personen möjlighet att använda stora ordförråd för att uttrycka sig.
En fördel är att det talade meddelandet underlättar för personen att få uppmärksamhet jämfört med om denne endast pekar på en bild. Talet kan dessutom hjälpa personen att koppla ord och bild. Å andra sidan kan det vara svårare för omgivningen att tolka om personen ofta oavsiktligt klickar på meddelanden. Det är viktigt att komplettera talande hjälpmedel med kommunikationskartor och kommunikationsböcker eftersom teknik kan krångla eller inte fungerar i alla situationer så som i badet.
Det verkar också vara individuellt om personen föredrar talande hjälpmedel eller kartor och böcker och vilket sätt som går snabbast att lära sig att använda. Det är viktigt att vara medveten om att det kan ta tid att lära sig nya kommunikationssätt och att många behöver utforska och prova sig fram under en lång period. Även enklare talande hjälpmedel som talknappar är användbara för att personen enkelt ska kunna göra sin röst hörd i olika aktiviteter.
Kommunikationspass
Kommunikationspass är ett kommunikativt stöd som hjälper omgivningen att kommunicera bättre med personen med Rett syndrom. Det beskriver i text och bild bland annat personen, hur personen kommunicerar och hur andra kan göra för att kommunikationen ska fungera som bäst. Om ett digitalt kommunikationspass används (exempelvis en Powerpoint) går det även att lägga in videoklipp. Det finns även som en app: RättVisat – Bräcke Innovation (brackediakoni.se)
Att få tillgång till kommunikationshjälpmedel
Kommunikationshjälpmedel förskrivs oftast av habiliteringen. Det finns en stor mängd olika kommunikationshjälpmedel och hur det exakt går till och vilka hjälpmedel som är möjliga att få varierar över landet.
Att utveckla språk och symbolkommunikation
Oberoende av vilken form av stort ordförråd personen har är strategierna som omgivningen kan använda för att stötta utvecklingen till största delen desamma. En mycket viktig uppgift för omgivningen är att personens AKK-system (AKK = Alternativ och kompletterade kommunikation) är tillgängligt i alla situationer. Om personen exempelvis har ett talande hjälpmedel kan det behövas en kommunikationsbok som backup, om batteriet tar slut eller om en kommunikationsbok av andra anledningar passar bättre att använda.
Ett annat viktigt sätt att lära ut att använda bilder som språk är att själv peka på bilder tillsammans med talet (pekprata eller modellera). På så sätt ges rikligt med möjligheter att koppla en bild till dess betydelse och hur den kan användas för att uttrycka behov, tankar och känslor. Om omgivningen använder bilder tillsammans med talet kan det också ge möjlighet till delad uppmärksamhet, t.ex. om personen tittar på bilden när samspelspartnern pekar på den. Det hjälper omgivningen att sakta ner taltempot och förtydliga viktiga ord i meningen genom att peka på dem. Det är dock viktigt att ha i åtanke att personens alla sätt att uttrycka sig är av värde. Bildsymboler kan också underlätta att förstå andras talade språk.
Mer information om kommunikativt stöd:
Musik påverkar oss alla fysiskt och känslomässigt. Inte minst spelar musik en viktig roll för personer med Rett syndrom. Rytmer gör att vi vill röra oss och klanger och harmonier väcker känslor och stärker vår uppmärksamhet. Musik kan både ge energi och verka lugnande.
Musikterapi kan ge stöd för personer med Rett syndrom att uttrycka känslor, visa olika förmågor och samspela med andra. Tillsammans med andra typer av insatser kan musikterapi ge stöd vid bedömning och utvärdering. Många praktiker och forskare lyfter särskilt upp musikens potential att motivera och engagera personer med Rett syndrom att vilja uttrycka sig. Musiken fyller en funktion som stödjande ramverk och kan underlätta för personen med Rett syndrom att samspela, ta initiativ och utföra rörelser och ljud på ett tillåtande och kravlöst sätt.
Kommunikativ musikalitet
Musik har en unik funktion att sammanföra människor. Det är inte för inte som vi människor i alla kulturer, sedan urminnes tider, har använt musik och andra konstnärliga uttrycksformer för att vara tillsammans och dela känslor och upplevelser.
Spädbarnsforskare och musikterapeuter menar att de allra första kontakterna mellan spädbarn och deras vårdnadshavare har en musikalisk struktur, där man kan känna igen element som puls, rytm och melodi i det dynamiska samspelet. Spädbarn och vårdnadshavare anpassar sig automatiskt efter varandras rytm och känsloläge. Det här kallas ibland för kommunikativ musikalitet och är något som sker automatiskt i mänskligt samspel från födseln.
Forskare med fokus på musikterapi och Rett syndrom lyfter att den medfödda musikaliteten även finns hos personer med Rett syndrom. De menar vidare att för personer som har mycket begränsat eller inget talat språk, så kan musiksamspel ge personen utökade eller alternativa möjligheter att uttrycka sig. Forskning visar att musik väcker ett stort intresse och ofta leder till kommunikativa initiativ och reaktioner från personer med Rett syndrom. Målgruppen har annars ofta svårt att upprätthålla och reglera vakenhet och engagemang, och många tar få initiativ till samspel, där också fysiska svårigheter så som dyspraxi försvårar rörelser. Flera forskningsstudier har visat att musikterapi kan stödja engagemang och uppmärksamhet, utveckla kommunikation och samspel, motivera till ökad handanvändning samt stödja känslomässigt uttryck hos personer med Rett syndrom och andra liknande diagnoser. Nedan ges några exempel på forskning om musikterapi tillsammans med personer med Rett syndrom.
Musikterapi – en väg till kontakt och utveckling för personer med Rett syndrom
Forskning visar att musikterapi kan spela en viktig roll för personer med Rett syndrom. Musik hjälper inte bara till att skapa lugn och trygghet i vardagen – den kan också locka fram förmågor som annars är svåra att upptäcka. Familjer berättar till exempel hur favoritmusik fungerar som ett verktyg för att trösta, minska oro eller underlätta kontakt. Studier har visat att även personer med mycket begränsad kommunikationsförmåga kan göra aktiva val, till exempel genom att välja sånger med hjälp av bilder. Genom musikterapi har barn och unga med Rett syndrom visat att de kan lära sig, utveckla samspel med andra och bli mer engagerade. En studie visade också att multisensoriskt musikdrama (där musiksamspel kombineras med sinnesupplevelser i en berättelse) ökade både barnets engagemang och lärarens förståelse för hur man bäst stöttar barnet. Rörelse och beröring hjälpte barnet att slappna av. Forskning har dessutom visat att musik påverkar hjärnan och kroppen på olika sätt –vilket bekräftar musikens kraft att både lugna och väcka sinnena. Sammanfattningsvis tyder forskningen på att musik förutom att bidra till glädje och engagemang för personer med Rett syndrom också kan vara ett viktigt stöd för lärande, kommunikation och välbefinnande.
Nedan ges tips på musikaktiviteter att göra i hemmet eller i er verksamhet. Följande strategier kan vara gynnsamma att tänka på i musikaktiviteter med personer med Rett syndrom:
Tips på musikaktiviteter och material:
Symtom och behandling
Att andas är en komplex kroppsfunktion som vi sällan tänker på förrän vi har problem med den. Andningen behövs för att tillföra syre till kroppen och föra bort koldioxid. Avvikande andningsmönster är mycket vanligt vid Rett syndrom.
Vid andning syresätts blodet och koldioxid vädras ut vilket är grundläggande för att vi människor ska må bra och fungera. Vid Rett syndrom är det vanligt med andningsproblem som har central orsak, det vill säga avvikande funktion i andningscentrum som har nära koppling till det autonoma nervsystemet. Vid cirka två års ålder kan perioder med avvikande andning under vaken tid observeras hos de flesta personer med Rett syndrom.
Det finns tydliga skillnader i andningsreglering över dygnet. Andningsstörningarna förekommer oftare under vakenhet än under sömn. Olika typer av avvikande andningsmönster kan uppkomma hos en och samma person och kan också ändra sig från dag till dag. Andningsavvikelserna kopplas till dysfunktion i den autonoma nervsystem och hjärnstammen vilket är vanligt hos personer med Rett syndrom. Tillsammans med avvikande andning kan svängningar i hjärtfrekvens och blodtryck förekomma vilket kan orsaka yrsel, oro, ångest och panikkänslor.
Det mest synliga andningsmönstret är episoder med hyperventilation (flåsande andning) som oftast uppkommer spontant, ibland i samband med emotionella situationer. Oftast följs detta av apné eller en period med ytlig eller långsam andning. Ytlig eller långsam andning kan uppkomma även utan hyperventilering. Hos en del personer förekommer också en särskild typ av andning: valsalva-andning som innebär att pressa ut luft med stängda stämband som vid krystning. Det resulterar i att en del av luften pressas ner i magsäcken och tarmen. I enstaka fall kan det leda till allvarliga tillstånd där magsäck eller tarmar skadas (ventrikel- eller tarmperforation). Hos personer med Rett syndrom kan också en apneustisk andning observeras. Denna typ av andning karakteriseras av djup, flämtande andning som avbryts, främst vid full inandning, följt av en kort, otillräcklig utandning.
Hos en del personer med Rett syndrom kan även andningsproblem under sömnen uppkomma med sömnapné och andningssvikt som kan behöva behandlas med ventilator (CPAP eller BiPAP). Detta förekommer oftare högre upp i åldrarna.
I en studie som publicerades 2018 följdes en stor grupp personer med Rett syndrom med årliga besök under flera år. Studien visade att nästan samtliga personer med klassisk Rett syndrom hade andningsproblem någon gång i livet. Personer med atypisk Rett syndrom och svår symtombild hade i stort sett lika ofta andningsproblem som personer med klassisk Rett syndrom medan personer med atypisk Rett syndrom och mildare symtom mer sällan hade andningssvårigheter (60% – 70%).
I denna studie klassificerades andningsdysfunktion som frekvent (allvarlig) om besvär i form av hyperventilering eller andningshållning (apnéer, apneustisk andning och Valsalvaandning) rapporterades vid mer än hälften av besöken och tydligt negativt påverkade normal daglig aktivitet. Hos personer med klassisk Rett syndrom rapporterades hyperventilation hos 54% till 68%, andningshållning hos 77% till 84%, luftsväljning hos 49% till 63% och luftpuffning (samla luft i munnen vid utandning och sedan släppa ut den) 49% till 68%. Frekvent (allvarlig) hyperventilation samt andningshållning rapporterades hos mindre än 40%.
Hur vanligt hyperventilering och andningshållning är varierar beroende på ålder och uppnår maximum vid 6 till 11 års ålder.
Vid klassisk Rett syndrom kan hyperventilering eller att hålla andan uppkomma eller återkomma som symtom i alla åldrar.
Avvikande andning kan påverka gasutbytet i luftvägarna och leda till för hög eller för låg koldioxidnivå. Risken att denna obalans blir allvarligt är liten även om andningsproblem är frekventa. Vid en snabbt tilltagande skolios kan dock finnas risk för snabbt stigande koldioxidnivå i blodet vilket behöver bevakas.
Personer med Rett syndrom kan även ha andningssvårigheter av andra orsaker, exempelvis på grund av låg grad av fysisk aktivitet, att formen av bröstkorgen är påverkad av skolios, ökad slembildning eller mediciner (t.ex. morfinpreparat eller bensodiazepinpreparat) som kan påverka andningen. Nattliga andningsuppehåll under sömn, så kallade sömnapnéer kan förekomma. De orsakas ofta av hinder i luftflödet genom luftvägarna (t.ex. förstorade halsmandlar, slappa och sammanfallande svalgväggar och att tungan faller bakåt mot svalgväggen). Snarkningar kan vara en varningssignal för sömnapnéer.
I handboken Kvalificerad omvårdnad i vardagen kan du läsa mer om andning och behandling generellt vid flerfunktionsnedsättning.
Bedömning och behandling
Föräldrar till barn med Rett syndrom behöver känna till att avvikande andningsmönster är mycket vanligt och att det kan påverka hjärtrytmen
Personer med Rett syndrom behöver regelbundet följas upp av specialister, som barnläkare, neurologer och eventuellt även pulmonologer.
Trots många år med intensiva studier för att kartlägga orsaken till, och framför allt mekanismen bakom andningsbesvär hos personer med Rett syndrom är detta fortfarande oklart. Det finns inte heller några effektiva behandlingar utprovade i större studier. Små studier har dokumenterat förbättring av andningsfunktion till exempel vid användning av ketogen kost för behandling. Det finns också rapporter om positiva effekter av behandling med buspiron, fluoxetin, topiramat men det visar sig vara mycket individuellt hur personer med Rett syndrom svarar på detta. Vid hyperventilation och Valsalva andning kan ökad fysisk aktivitet ha betydelse. Vid stora problem med luftsväljande används hos en del personer gastrostomi för att lufta magen med bra effekt.
Behandlingen av nattliga sömnapnéer riktas in på att förbättra andningen under natten och detta kan se olika ut beroende på orsaken. Ibland räcker det med att få en bättre position under sömnen, vid gastroesophageal reflux kan antirefluxmedicin hjälpa, ibland behöver halsmandlarna opereras bort. Vissa kan ha hjälp av en speciell apnébettskena eller sömntandställning. En del behöver få hjälp av olika apparater som stödjer andningen genom att ge ett kontinuerligt positivt lufttryck vid andning (CPAP, BIPAP). Personer med centrala sömnapnéer behöver mer avancerad hjälp i form av en respirator som tar över styrningen av andningen under sömn.
Hos personer med flerfunktionsnedsättning kan det ta betydligt längre tid att lära sig få kontroll över blåsan och tarmtömningen och inkontinens (läckage av urin eller avföring) och enures (urinläckage under natten) är vanligt. En del personer kan lära sig kontrollera avföring men inte blåsan.
Inkontinens är väldigt lite beskrivet i forskningen om Rett syndrom trots att både urin- och avföringsinkontinens är mycket vanligt. I en studie av Giesbers och kollegor förekom urininkontinens både dag och nattetid hos nästan alla och avföringsinkontinens rapporterades hos 72% av personerna med Rett syndrom under dagtid och hos 57% av personerna under nattetid. Inkontinens kunde inte kopplas till ålder och nivå av adaptiv funktion. Tömningen av blåsan kan vara ineffektiv och leda till infektioner i urinvägarna.
Förutom svårigheter med att känna reflexen och lära sig kontrollera den kan det finnas andra orsaker till inkontinens. Avföringsinkontinens kan orsakas till exempel av förstoppning (förstoppningsdiarréer när vattentunn avföring rinner på sidan av stora, hårda klumpar) eller vid diarréer. Urinläckage kan orsakas av till exempel infektioner samt olika typer av missbildningar i urinvägarna.
Vid diagnostik kan olika undersökningar användas för att identifiera orsaken till inkontinensen, till exempel bukultraljud, en 48 timmars blåsdagbok, röntgenundersökning av buken (buköversikt) och mätning av urinflöde. En blåsdagbok kan underlätta att hitta bra rutiner för blåstömning. I blåsdagboken noteras bland annat noggrant intag av dryck samt hur ofta och hur stor mängd personen har kissat.
Behandling
En viktig form av behandling är att vid behov undanröja medicinska orsaker som förstoppning eller infektioner. Utöver detta har individuellt anpassad, intensiv toaträning haft goda resultat i andra grupper med flerfunktionsnedsättning. Sådan toaträning inleds med en noggrann kartläggning av till exempel hur, när och var personen behöver gå på toaletten i olika miljöer. Därefter gör man med stöd av exempelvis habiliteringen en plan för att genomföra och utvärdera den systematiska toaträningen. Endast schemalagda toalettider verkar inte vara effektivt vid Rett syndrom.
I Podden Funka Olika från Habilitering och hälsa tas toaträning upp i två avsnitt.
Enligt en studie med nästan 1000 personer med RS i olika åldrar (småbarnsålder till 30+) tog del, rapporteras mag-tarmbesvär hos upp till 92% personer.
Gastroesofageal reflux
I en stor studie rapporteras gastroesofageal reflux hos 39% personer med Rett syndrom. Det är ungefär samma förekomst som generellt bland personer med flerfunktionsnedsättning. Det finns en del enkla åtgärder att prova som kan lindra besvären som beskrivs nedan.
Gastroesofageal reflux (GER) är ett tillstånd när surt magsäcksinnehåll kommer upp till matstrupen (esofagus). Detta händer framför allt efter måltider, när personen böjer sig eller böjs framåt, vid lyft samt i liggande position. Förutom irritation och obehag kan det leda till inflammation i matstrupen.
Upprepade GER-episoder kan övergå till ett kroniskt tillstånd som kallas för gastroesofageal reflux-sjukdom eller GERD.
De vanligaste symtomen är sura uppstötningar och halsbränna (brännande smärta i mitten av bröstet). Sämre aptit eller matvägran samt sväljningsbesvär och kräkningar kan också vara tecken på GER. Nattliga reflux-episoder kan leda till sömnproblem. GER kan påverka luftvägarna. Surt magsäcksinnehåll kan exempelvis irritera luftvägarna och orsaka hosta, heshet och astmaliknande tillstånd. Uppstötningarna kan också komma in i luftvägarna och leda till lunginflammation. Det är också vanligt med emaljskador på tänderna. Vid långvarig reflux kan den sura vätskan leda till kronisk inflammation och förändringar i nedre delen av matstrupen och anemi kan uppkomma.
GER orsakas av dysfunktion i en muskel mellan magsäcken och matstrupen som kallas för övre magmunnen (lower esophageal sphincter). Den är en del av en stor muskel, diafragma, som skiljer brösthålan och bukhålan åt. Orsaken till dysfunktionen är inte klarlagd men viktiga generella riskfaktorer är låg muskeltonus samt när trycket i bukhålan ökas såsom vid luftsväljande, skolios eller fetma. Både låg muskeltonus, förstoppning, luftsväljande och även skolios uppkommer ofta vid Rett syndrom. Dessutom kan peristaltiken vara långsammare som en effekt av autonom dysfunktion vilket också är vanligt vid Rett syndrom.
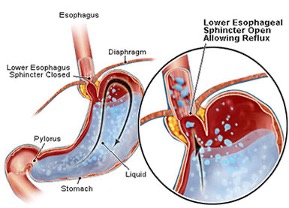
Bilden från eNetMD visar sur reflux genom öppet övre magmunnen (lower esophageal sphincter).
För stora matportioner, kan leda till reflux-symtom exempelvis hos personer som har nedsatt mättnadskänsla eller som får mat via sond eller stomi. Detsamma gäller om det finns hinder i passagen från magsäcken ner till tarmen. En del mediciner, t.ex. vissa smärtlindrande tabletter eller antiepileptiska läkemedel samt fettrik eller starkt kryddad mat, kaffe och juice kan också öka risken för reflux-symtom.
Hos de flesta personer med Rett syndrom ställs diagnosen utifrån observation och information från närstående. Om symtomen minskar till följd av en provbehandling kan det också vara till hjälp vid diagnostisering. En säker diagnos kan ställas med en kontraströntgenundersökning av matstrupen och magsäcken.
Vid mer komplicerade situationer (t.ex. blodiga kräkningar) kan en mer avancerad undersökning – gastroskopi – göras, vilket oftast kräver narkos.
En annan undersökning kallad 24 timmars pH-metri, där surheten i matstrupen mäts med sond, utförs framför allt på personer som fortsätter att ha svåra besvär trots farmakologisk behandling och som därför ska opereras.
Behandling av GER
Behandling behöver inte alltid vara svår och avancerad. Några enklare åtgärder som kan underlätta är:
- Regelbundna måltider och att hellre äta små portioner ofta än att äta stora portioner sällan
- Att äta i sittande position och att sitta eller helst stå och gå mellan måltiderna underlättar och förbättrar matpassagen. Att undvika att äta strax innan sänggående, att höja sängen huvudändan (ca 30 – 40 cm) samt att helst ligga på vänster sida minskar risken för problem nattetid.
- Receptfria mediciner kan vid behov lindra symtomen ganska snabbt. Vanliga alternativ är syraneutraliserande medel (Samarinpulver) eller slemhinnetäckande medel, t.ex. Gaviscon. Effekten av denna behandling är kortvarig och det är viktigt att kontrollera informationen på bipacksedeln för eventuella kontraindikationer om personen behandlas med andra läkemedel.
Om dessa åtgärder inte hjälper kan behandling med läkemedel, så kallade protonpumpshämmare (t.ex. Omeprazol eller Esomeprazol) eller H2-blockerare (Famotidin, Pepcid) läggas till. Dessa mediciner är också receptfria men det är bäst att diskutera behandlingen med en läkare som kan bestämma dosen och också följa effekterna av behandlingen. Vid svåra besvär är även operativ behandling motiverad.
Läs också Halsbränna – magsaftreflux på 1177.se samt handboken Kvalificerad omvårdnad i vardagen av Ann-Kristin Öhlund som täcker många områden inklusive mag- och tarmbesvär.
Förstoppning/diarréer
Förstoppningsproblematik rapporteras hos cirka 80% av personerna med Rett syndrom. Diarréer observeras ganska sällan och är oftast effekten av matintolerans/matförgiftning, infektion eller biverkning av medicinering.
Förstoppning innebär att tarmen töms mer sällan än ca tre ggr per vecka. Hur ofta man behöver gå på toaletten är individuellt men det bör inte ske mer sällan än varannan till var tredje dag. Problem med förstoppning förekommer i alla åldrar och är mycket vanligt bland personer med neurologisk funktionsnedsättning men kan även drabba personer utan neurologisk diagnos.
Hos personer med Rett syndrom bidrar långsammare passage genom tarmarna (dysmotilitet) till att förstoppning uppstår.
Följande faktorer kan öka risken för förstoppning:
- för lite vätska
- för lite fibrer
- för lite rörelse
- förändring i matvanor
- resor eller byte av miljöer
- stress
Förstoppning kan också vara en biverkning av olika läkemedel som används vid behandling av andra tillstånd, tex smärta eller blodbrist. Smärtlindrande mediciner som innehåller morfin eller andra opioider såsom Tramadol eller olika järnpreparat kan ofta leda till problem med tarmtömning. Långvariga problem med förstoppning kan också vara tecken på sjukdom, till exempel Hirschsprungs sjukdom, IBS (Irritable Bowel Syndrome), hypothyreos eller celiaki.
Förutom att tarmen töms glesare än var tredje dag är avföringen oftast också hård och trög att få ut. Det kan göra ont när tarmen töms och sprickor kan uppkomma i öppningen av ändtarmen vilket ytterligare kan förvärra förstoppningen.
Avvikande andningsmönster hos personer med Rett syndrom med hyperventilation, andningshållande och luftssväljning (aerofagi) leder till stora problem med uppspänd mage vilket rapporteras hos 53%-70%. Problem med uppspänd mage kan variera under tiden och påverkas av faktorer som stress och oro. Detta leder oftast till smärta som ytterligare eskalerar stress och oro. Förstoppning kan ytterligare förvärra situationen. Tillståndet är svårt att behandla eftersom det inte finns någon effektiv behandling mot avvikande andningsmönster. Gasbildande medel kan vara till hjälp. I vissa fall kan gastrostomi ge en positiv effekt. Tillståndet är viktigt att bevaka eftersom det i enstaka fall har lett till allvarlig komplikation i form av ventrikelperforation.
Ett annat tillstånd som är sällsynt vid Rett syndrom (uppkommer hos ca 4,4%) men som ibland kan leda till allvarliga konsekvenser är gallvägssjukdom. Symtomen i form av återkommande buksmärtor lokaliserade under höger revben, irritabilitet, viktminskning och kräkningar är ganska ospecifika och svåra att tolka hos personer som inte själva kan förmedla sig eller kanske uppfatta exakt var i kroppen det gör ont. Nedsatt peristaltik i gallvägarna som ses hos personer med Rett syndrom och låg fysisk aktivitet är faktorer som bidrar till gallvägssjukdom. Blodprover kan i 50% av fallen vara normala. Diagnosen kan ställas med ultraljud av buken men ofta behövs mer avancerad radiologisk diagnostik för att ta beslut angående behov av kirurgisk behandling.
Förstoppning leder ofta till nedsatt aptit, illamående, gastroesofageal reflux samt kräkningar. Detta kan i sin tur minska vätskeintaget, orsaka allmän trötthet samt påverka fysisk aktivitet negativt. Personen kan alltså hamna i en negativ spiral och problemen med tarmtömning bli allt större.
Långvariga problem med förstoppning kan också försämra nutritionen, leda till ökad trötthet och feber samt öka krampfrekvensen hos personer med epilepsi. Stora, hårda avföringsklumpar som blir liggande i sista delen av tjocktarmen och ändtarmen kan påverka urinblåsan och medföra svårigheter att tömma blåsan, framkalla täta trängningar och leda till reflux i urinledarna. Hos personer med spasticitet kan denna öka vid förstoppning.
För att kunna upptäcka förstoppning hos personer med Rett syndrom är det viktigt att ha noggrann kontroll över avföringsfrekvens, mängd, färg och konsistens. Det är bra att notera detta i en almanacka och eventuellt även ha någon typ av pott-/bajskalender. För att standardisera informationen är det bra att använda en särskild skala som heter Bristolskalan (Bristol Stool Chart) för att beskriva färg och konsistens på avföringen.
Det kan vara svårt att påvisa att det rör sig om förstoppning exempelvis vid förstoppningsdiarréer. Brunt avföringsfärgat vatten och lösare tarminnehåll kommer då förbi avföringsklumpar i tjocktarmen. Personen kan då ha avföring varje dag, ibland flera ggr per dag men inte så stor och avföringen brukar dessutom vara lös.
Information från vårdnadshavare/assistenter bör kompletteras med en noggrann fysisk bukundersökning och ändtarmundersökning med finger ibland kompletterad med röntgenundersökning av magen – så kallad buköversikt (BÖS). I sällsynta fall bör undersökningen även kompletteras med rektoskopi (undersökning med ett tunt plaströr med en liten kamera som införs i ändtarmen).
Vid misstanke om eventuella bakomliggande sjukdomar bör blod och/eller avföringsprover tas samt tjocktarmen undersökas med koloskopi (undersökning liknar rektoskopi men kameran införs högre upp till tjocktarmen).
Förebyggande behandling
Vid misstänkt förstoppning är det viktigt att reagera snabbt. Tillräcklig stort vätskeintag (se tabell), fiberrikkost och fysisk aktivitet har den största betydelsen. Till vätskeintaget räknas även all vätska som kommer med mat.
| KROPPSVIKT | VÄTSKEBEHOV UNDER 24 TIMMAR |
|---|---|
| Under 5 kg | 150 ml per kg |
| 5-10 kg | 100 ml per kg |
| 11-20 kg | 1000 ml + 50 ml för varje kg över 10 kg |
| Över 20 kg | 1500 ml + 20 ml för varje kg över 20 kg |
| Vuxna över 50 kg | Minst 30 ml/kg kroppsvikt; max 2000 ml |
När det gäller kost är det viktigt med frukter och grönsaker samt fullkornsprodukter, grovt bröd med mera. Katrinplommon, aprikoser, russin, fikon och päron samt linfrön har laxerande verkan. Viktigt att komma ihåg är att fiberrik kost ökar vätskebehovet. Kosten bör också innehålla tillräckligt mycket fett. Probiotika, bakteriekultur som är gynnsam för tarmen, kan också vara bra. Det är bra att kontakta dietist för att få råd och förslag angående både kostinnehåll samt konsistens och även hjälp med bedömning av kalori- och vätskebehov.
Tarmmotorik aktiveras också av matintag. Det är därför bra att planera in och skapa rutin för toalettbesök efter en och samma måltid varje dag. Det bör finnas tillräckligt med tid för toalettbesök och det får inte bli stressande. Det är också viktigt att se till att personen känner sig lugn, trygg och avslappnad på toaletten. En adekvat, bekväm position på toalettstolen, med stabilt sittande, lätt framåtlutad bål samt stöd för armarna och fötterna underlättar tarmtömning. Det kan också behövas någon lockande sysselsättning för personer som behöver sitta länge på toaletten.
Fysisk aktivitet, vilket även inkluderar stående eller passiva rörelser, stimulerar tarmmotorik och underlättar avföringspassagen i tarmen. Massage och taktil stimulering kan också vara till hjälp ibland.
Om dessa förebyggande procedurer och anpassningar inte är tillräckliga kan tarmreglerande – volymökande medel som tex Movicol eller Laktulos användas. Dessa läkemedel samt andra naturbaserade preparat är receptfria. Det finns inte något universalmedel och det är viktigt att testa olika för att hitta den som passar bäst. Detta gäller framför allt effekten men också mängd och form. Det är också bra att ha kontakt med en speciellt utbildad sjuksköterska som kallas tarmsjuksköterska eller uro-tarmterapeut. De kan tipsa om olika tarmreglerande medel samt lära vårdnadshavare eller assistenter att tömma tarmen med hjälp av lavemang. Vid lavemang ges mindre (Resulax) eller större (Klyx) mängd läkemedel som har återfuktande och glidande effekt direkt i ändtarmen. Ibland kan även lavemang med blandning av matolja och vatten ges. Hos små barn kan mikrolavemang eller stolpiller användas.
När stora mängder hård avföring samlas i tarmen kan stora avföringsklumpar bildas, så kallade fecalom. Detta tillstånd behöver behandlas i kontakt med sjukvård. I dessa fall görs i första hand behandlingsförsök med stora doser av Movikol samt tarmstimulerande läkemedel (Laxoberol, Dulcolax) och upprepade lavemang under 4 – 5 dagar. Om detta inte hjälper måste tarmtömning ske i narkos. Om bakomliggande sjukdom orsakar förstoppningen måste den också behandlas.
Epilepsi är vanligt vid Rett syndrom och därför är det viktigt att vara uppmärksam på tecken på anfall. Epilepsi innebär upprepade, oprovocerade, okontrollerade störningar eller utbrott av hjärnaktivitet som kallas epileptiska anfall. Medvetandet är ofta mer eller mindre påverkat under anfallet. En del personer kan ha ett par anfall under hela livet medan andra kan ha flera anfall per dag och behöva behandling livet ut.
Hur anfallen ser ut beror på var i hjärnan de uppstår (se anfallstyper). Anfall uppkommer oftast plötsligt och kan vara i sekunder, ibland minuter, och slutar oftast av sig själv.
De flesta personer med Rett syndrom har någon gång under livet krampanfall. Hur vanligt epilepsi är varierar mellan olika studier (från 30%-82%). Det finns ingen klarlagd koppling mellan typ av mutation i MECP2 genen och hur ofta eller svåra anfall en person har. Däremot finns en koppling till ålder. Epilepsi är vanligast mellan 3 och 5 års ålder och förekommer före 8 års ålder hos >80 %. Vid krampdebut innan två års ålder är det vanligt att epilepsin är svårbehandlad och fler anfallstyper förekommer hos en och samma person. Anfallsdebut efter fem års ålder brukar ses som en prognostiskt bra faktor. Med åren blir anfallen oftast färre och en del vuxna kan även vara anfallsfria även utan krampmedicinering.
Epilepsi kan påverka vardagslivet både för personen som har epilepsi och deras familjer. Hur stor påverkan blir beror på när epilepsin debuterar, personens allmänna hälsotillstånd, typ av anfall, svar på behandling m.m. Infektioner, feber, sömnbrist, stress, smärta m.m. kan sänka kramptröskeln och leda till flera anfall. Hormonförändringar vid puberteten hos flickor kan orsaka att epilepsi utbryter eller att epilepsibilden ändras både med fler anfall och färre. Hos en del personer med epilepsi finns samband mellan anfallsfrekvens och faser i menstruationscykeln. Olika miljöfaktorer såsom blinkande ljus eller värme kan också framkalla anfall vilket behöver beaktas när aktiviteter planeras.
Anfallstyper
De epileptiska anfallen kan se olika ut. Det är vanligast att en person endast har en typ av anfall men en del personer har flera olika typer av anfall. Typ av anfall hos en person kan förändras över tid. Beroende på var anfallet startar delas de in i generaliserade anfall, fokala anfall och anfall med okänd start.
Anfallstyper vid Rett syndrom
De epileptiska anfallen kan se olika ut. Det är vanligast att en person endast har en typ av anfall men cirka en fjärdedel av personerna med Rett syndrom och epilepsi har två eller flera olika typer av anfall. De vanligaste anfallstyperna är fokala anfall med eller utan sekundär generalisering samt generaliserade toniskkloniska anfall. Dessa båda anfallstyper ses hos cirka hälften av personerna med Rett syndrom, medan frånvaroanfall ses hos cirka 15%. Andra typer av anfall som myokloniska anfall och atoniska anfall rapporteras betydligt mer sällan.
Det är också vanligt med episoder och beteenden som inte har epileptisk bakgrund men som kan vara svåra att skilja från epileptiska anfall. Några exempel är plötsliga episoder med skratt, avvikande andning, snabb eller långsam hjärtrytm, svettningar och hastig rodnad (flush) i ansikte och ibland även skakningar eller ryckningar i muskler, förlust av muskelspänning, vidgade pupiller eller stirrande blick. Dessa episoder kallas för paroxysmala icke epileptiska manifestationer och beror på dysfunktion i hjärnstammen. Studier visar att dessa icke-epileptiska episoder är svåra att skilja ifrån epileptiska anfall, även för föräldrar.
Diagnos
För att avgöra om en anfallsepisod har epileptisk bakgrund är det mycket viktigt med så omfattande information som möjligt från de som var närvarande vid anfallen, exempelvis:
Det är också bra med en videoinspelning av anfallen. Utifrån ovanstående information samt kännedom om patientens tidigare symtom tas beslut om vidare diagnostik. Diagnosen epilepsi ställs oftast först efter två oprovocerade anfall (inte utlösta av exempelvis akut feber). Om det finns en känd genetisk diagnos med ökad risk för epilepsi såsom Rett syndrom räcker det med ett anfall för att ställa diagnosen epilepsi och epilepsibehandling behöver då oftast sättas in.
Vid misstänkt epilepsi görs en neurologisk undersökning och en undersökning som kallas EEG (elektroencefalografi). Läs på 1177 hur en EEG-undersökning går till. Dessa kompletteras med blodprover och ofta en röntgenundersökning av hjärnan (datortomografi eller magnetisk resonans) för att kartlägga orsaken till epilepsin.
Om det finns misstanke att personens anfall kan ha icke-epileptisk karaktär är det viktigt att försöka dokumentera dessa under pågående långtids video-EEG. En och samma person kan ha både epileptiska kramper samt icke epileptiska paroxysmer. Även med långtids video-EEG är diagnostiken av epilepsi svår vid Rett syndrom eftersom de flesta personerna med syndromet har avvikande EEG-kurva efter ca 3 års ålder. Det ses en del avvikande förändringar som uppkommer oberoende av anfall. De avvikande EEG-mönstren kan även ändras med åldern.
Behandling och åtgärder
Epilepsimedicinering, särskilt om det behövs flera preparat, kan leda till en del begränsningar och biverkningar som behöver beaktas och kan påverka personens livskvalitet.
Individens prestationsförmåga och dagsform kan påverkas och planerade aktiviteter behöver därför kanske anpassas eller ändras. Detta är särskilt viktigt om personen har flera anfall under ett dygn. Anfall under natten påverkar längden och kvaliteten på sömn. Assistenter och andra i omgivningen som träffar personen med epilepsi regelbundet behöver få information om personens epilepsi, typ av anfall och vad de ska göra vid eventuella anfall. Vid vissa aktiviteter är det nödvändigt att ha dubbel personal av säkerhetsskäl.
Behandling vid akut anfall
Första hjälpen som ges vid ett anfall är densamma oavsett om personen haft tidigare anfall eller inte. Personen ska placeras i ett bekvämt och säkert läge, helst liggande på golvet för att minska risken för fall. Det är viktigt att ta bort saker runt omkring, ta bort glasögon och lossa slips och skärp samt lägga någonting mjukt under huvudet.
För att undvika tandskada ska ingenting sättas mellan tänderna. Personen bör inte hållas fast men ska inte lämnas ensam under pågående anfall. Om det finns någon i omgivningen som kan filma anfallet är det värdefullt för att kunna visa hur anfallet såg ut för personal på akuten eller för epilepsisjuksköterska/läkare.
De allra flesta epileptiska anfall går över av sig själv efter 1 till 3 minuter så det är viktigt att försöka avvakta och vara lugn. Efter att anfallet gått över ska personen läggas på sidan i en säker position men bör inte lämnas ensam förrän han eller hon återhämtar sig helt. Det är viktigt att luftvägarna är fria och att personen andas fritt. Det är också bra att se till att personen är skyddad mot fall och ligger på mjukt underlag om ett nytt anfall skulle uppkomma.
Om personen har en känd epilepsi brukar denne ha med sig medicin som kan ges vid akuta tillfällen av förälder, assistent eller annan personal med kunskap om hur den ska användas (så kallad delegering). Medicinen är flytande och kan ges i kinden (Midazolam) eller i ändtarmen (Diazepam). Medicinen har muskelavslappnande och, för hjärnan, lugnande effekt.
Ambulans bör tillkallas (112) när det är oklart om anfallet är epileptiskt, om det är det första anfallet personen har, om anfallet inte går över efter 5 minuter eller vid snabbt upprepade anfall utan att personen blir helt återställd emellan. Detsamma gäller om personen har skadat sig under anfallet.
Anfall som pågår längre än 20 till 30 minuter, eller kommer tätt efter varandra under ca 30 minuter utan att personen återhämtar sig emellan, kallas för status epileptikus. Behandling av detta tillstånd sker på sjukhus och oftast på intensivvårdsavdelning med intravenös kramplösande medicin.
Långsiktig behandling
Epilepsibehandling startas av neurolog eller i samråd med denne. Det är viktigt att ha regelbunden läkaruppföljning och samarbete med epilepsisjuksköterska. Målet med behandling är att helt eller delvis förebygga att anfall uppkommer genom att minska nervcellernas känslighet, så kallad kramptröskel, och återställa hjärnans aktivitet.
Samtidigt är det viktigt att behandling ger minsta möjliga biverkningar. Det finns en stor mängd antiepileptiska läkemedel. Typ av epilepsi/anfall, kön, ålder, personens hälsotillstånd och eventuella andra mediciner har betydelse vid val av antiepileptisk behandling. Det finns inget stöd för att någon särskild medicin är bättre än någon annan mot epilepsi hos personer med Rett syndrom.
Bland personer med Rett syndrom som har epilepsi svarar drygt 50% av personer bra på behandling men ca 33 % har svårbehandlad epilepsi och behöver flera olika antileptika för att bli krampfria. Hos ca 5% av gruppen personer med Rett syndrom minskar inte kramperna vid behandling trots flera antiepileptiska mediciner. Det kan då bli aktuellt med behandling med ketogen kost eller Vaguservstimulator (VNS). I enstaka fall kan även kirurgisk behandling vara alternativ.
Ett fåtal personer kan ha sporadiska anfall och behöver då ingen medicinering. Med åren kan behov av medicinering minskas.
Det finns en del allmänna regler men ingen färdig mall eller ”kokbok” utan behandlingen anpassas individuellt och justeras utifrån effekt. När behandling sätts in eller ändras är det viktigt att noggrant notera frekvens på anfall, anfallens karaktär/mönster, tiden på dygnet när de uppkommer, justeringar av medicindos och faktorer som kan ha haft betydelse för att anfall uppkommer. Noteringarna kan göras i en anfallskalender (s.k. anfallsdagsbok) men det går även att notera i en vanlig almanacka. Det är också viktigt att notera eventuella biverkningar eller reaktioner som eventuellt skulle kunna vara en biverkning.
Biverkningar kan oftast uppstå när personen påbörjar behandlingen med epilepsimedicin eller höjer dosen. Exempel på biverkningar är allergiska reaktioner; oftast i form av hudutslag och klåda, illamående, diarréer, ont i magen, förändringar i blod- och/eller leverenzymvärden, yrsel, balansproblem, dåsighet och trötthet, lättretlighet och koncentrationsproblem.
Ibland måste behandlingen avbrytas men det kan även räcka att minska dosen och/eller att trappa upp medicinen långsammare. En del biverkningar såsom hormonstörning, viktökning/minskning, tillväxt av tandkött, akne, håravfall eller hårväxt på ovanliga ställen, minnes- och koncentrationssvårigheter, apati eller depression uppkommer först efter flera år. Det är viktigt att uppmärksamma detta och vid behov diskutera med den läkare som är ansvarig för epilepsibehandling för att hitta den bästa lösningen för individen.
Vid behandling med epilepsimediciner behöver prover ibland tas för att kontrollera koncentrationen av medicin i blodet. Det är särskilt viktigt i början av en behandling eller om effekten av behandlingen plötsligt minskar.
Olika faktorer kan sänka kramptröskeln och därmed leda till att anfall lättare uppkommer. Otillräckligt med sömn, feber, infektion, lågt blodsocker och hunger, blinkande ljus från TV-apparat eller dator samt stress är kända sådana faktorer.
Vissa ämnen som alkohol och droger samt en del mediciner som centralstimulantia kan också öka risken för anfall liksom om personen inte får sin epilepsimedicin. Det är alltså viktigt att undvika faktorer som sänker kramptröskeln.
I många fall räcker det att prova ett eller två läkemedel. Cirka 30% av de som har epilepsi behöver ta flera antiepileptiska mediciner samtidigt för att bli av med anfall eller reducera anfallens frekvens och intensitet. Detta kallas refraktär eller svårbehandlad/terapiresistent epilepsi. Det är då svårt att få balans mellan behandlingseffekten och biverkningarna och i många fall accepteras enstaka anfall för att få mindre biverkningar och bättre livskvalitet
Det finns exempelvis personer som behövt kirurgisk behandling eller som varit hjälpta av vagusnervstimulator (se nedan).
Alternativ epilepsibehandling
Om det är svårt att uppnå anfallsfrihet med den första eller andra medicinen som provas är det en indikation att överväga alternativ behandling. Vid indikation för alternativ epilepsibehandling bör ett regionalt specialistepilepsicenter kontaktas för vidare diskussion.
Kirurgi
Det finns flera kirurgiska behandlingsmetoder. En operation kan oftast ha bra resultat om det går att identifiera området i hjärnan som framkallar störningarna av impulser och leder till kliniska anfall. Området behöver även vara lättillgängligt för operation.
Inför en eventuell operation krävs en del relativt komplicerade undersökningar. Omfattningen av operationen behöver preciseras så noggrant som möjligt. Det är också viktigt att säkerställa att den del som planeras att bortopereras inte har någon viktig funktion.
Vid en viss typ av operation, så kallad callosotomi, delas bindningen mellan hjärnhalvorna (corpus callosum) totalt eller delvis för att minska spridning och generalisering av anfall. I sällsynta fall utförs lobotomi eller en hemisfärotomi när delar eller en hel hjärnhalva tas bort.
Ketogen kost
Ketogen kost är en annan alternativ behandlingsmetod vid svårbehandlad epilepsi. Vid klassisk ketogen kost innehåller måltiderna 90% fett, 6% proteiner och endast 4% kolhydrater. Denna diet behöver inledas successivt och följas noggrant och kontinuerligt av dietist.
Den används oftast under en begränsad tid och kan eventuellt ersättas med mindre restriktiva varianter (modifierad Atkins diet och Low glykemic index-diet). Även dessa varianter kräver samarbete med dietist och kontroller för att minska risken för biverkningar.
Vagusnervstimulering (VNS)
VNS är en behandling som kräver operation men den utförs inte inom det centrala nervsystemet. En liten, ca 3 cm i diameter stor dosa som fungerar som en pacemaker inplanteras under huden på vänster sida av bröstkorgen och kopplas ungefär i mitten av halsen till vagusnerven på samma sida med mikroskopiska trådar. Därigenom skickas sedan impulser som motverkar epileptiska störningar i hjärnan.
Symtom
Barn med Rett syndrom har en till synes normal motorisk utveckling den första tiden. Därefter inträffar ett utvecklingsstopp och en efterföljande regressionsfas med försämring av bland annat motoriska färdigheter. Det innebär att barnet kan ha lärt sig motoriska färdigheter som att krypa och gå när rörelserna plötsligt visar tecken på ataxi, som innebär att de blir ryckiga och mindre samordnade. Samtidigt minskar grundmuskelspänningen i kroppen vilket gör att barnet har svårare att hålla sin kropp upprätt. Den minskade kroppspänningen gör att kroppen sjunker ihop, balansen försämras och förmågan att uppfatta kroppens position i rummet blir nedsatt.. Denna regressionsfas när de tappar tidigare inlärda motoriska färdigheter inträffar mellan 1 – 4 års ålder och pågår från några veckor till månader.
Ett centralt symtom för Rett syndrom är nedsatt förmåga att planera, starta och genomföra viljestyrda rörelser – dyspraxi. Det innebär att reaktionen vid motorisk aktivitet kan vara fördröjd och att det kan ta lång tid för motorisk respons på instruktioner. Finmotoriken drabbas särskilt tydligt eftersom handfunktion kräver stor samordning. I stället för att använda händerna på ett meningsfullt sätt utvecklar många repetitiva handvridningar och gnuggningar. Individer med Rett syndrom har ofta påverkan på autonoma, icke viljemässiga nervsystemet med dysfunktion i andning, blodtryck, vakenhet, nedsatta tarmrörelser, påverkan på blodcirkulation med kalla fötter etc.
När barnet är mellan 2 – 10 år börjar de återfå motoriska funktioner som att viljemässigt kunna använda händerna, säga enstaka ord och utveckla förmågan att stå och gå även om balansen ofta är nedsatt. Hur mycket funktioner som återinlärs är helt individuellt. De motoriska svårigheterna varierar mycket mellan olika individer med Rett syndrom. Vissa kan gå självständigt (72 %) medan andra saknar förmåga att sitta, stå och förflytta sig.
Den generella muskelspänningen hos personer med Rett syndrom kan förändras över tid. I yngre åldrar är den oftast låg men med ålder kan den bli hög eller växlande i olika kroppsdelar. En hög muskelspänning eller spasticitet kan påverka elasticiteten i musklerna och leda till felställningar i rygg, höfter, fotleder och tår. Även låg muskelspänning kan leda till instabilitet och överrörlighet i leder vilket ökar risken för permanenta felställningar.
Risken för sekundära komplikationer är stor. Ungefär 70 % drabbas av skolios, hälften får felställningar i fötterna och höftledsubluxation (att höftleden delvis glider ur led), förekommer hos en tredjedel av personerna. Frakturer är fyra gånger vanligare hos personer med Rett syndrom jämfört med den övriga befolkningen. Troligen är det ett resultat av minskad bentäthet på grund av inaktivitet men kan också bero på brist på D-vitamin och kalcium eller biverkningar från epilepsimedicinering. Här är det viktigt för nätverket att vara observant på tecken på benbrott om personen själv inte kan kommunicera smärta och obehag.
Grovmotoriska förmågor kan försämras i vuxen ålder, men det är inget som sker hos alla. Försämringen kan vara åldersrelaterad eller bero på minskad fysisk aktivitet. Även om personer har förlorat motoriska förmågor finns det möjlighet att återfå funktioner igen. Det finns forskningsrapporter om personer med Rett syndrom som återfått förmågor i vuxen ålder exempelvis att resa sig upp från golv eller att gå efter att ha suttit i rullstol i många år.
Behandling
Motorisk bedömning och behandlingsråd
Fysioterapi är en central del i behandlingen av personer med Rett syndrom och syftar till att bevara, återerövra och utveckla motoriska funktioner samt minska risken för sekundära åkommor. Vissa funktioner kan återfås men ofta kvarstår förändrat muskeltonus, nedsättningar i koordination, planering och utförande av rörelser.
En fysioterapeut gör en motorisk bedömning och ger behandlingsråd utifrån personens aktuella behov och begränsningar. Insatserna kan innehålla allt från dagligt stående med hjälpmedel till repetitionsbaserad funktionell träning, balansträning, aktivering av händer, spasticitetsreducering, träning av muskelstyrka och ledrörlighet. En tydlig rekommendation för personer med Rett syndrom är att stående och gångträning är viktigt för att motverka negativa följder såsom felställningar i leder och rygg samt benskörhet. Genom att belasta skelett och aktivera muskler påverkas bland annat rörlighet, muskelstyrka, koordination, mag- och tarmfunktion samt hjärt- och kärlfunktion. Utöver de fysiska fördelarna bidrar ståendet även till ökad möjlighet till social interaktion och kommunikation, vilket är viktigt för det psykiska välbefinnandet.
Funktionell träning i vardagen
För god effekt bör den motoriska träningen förutom att vara individuellt anpassad också vara integrerad i vardagen och kunna genomföras och upprepas på ett enkelt sätt. Man bör utgå från personens egna intressen och behov för att utföra träningen så lustfylld som möjligt. Eftersom många individer med Rett syndrom har en begränsad förmåga att initiera motoriska aktiviteter själva, krävs ofta ett aktivt stöd från nätverket för att skapa möjlighet till rörelse och fysisk aktivitet.
Fysisk aktivitet rekommenderas
Här följer några förslag till fysiska aktiviteter för att stimulera motoriken för personer med Rett syndrom som har en begränsad motorisk förmåga:
Interventioner för att motverka felställningar i rygg och leder
Nedan följer vanliga felställningar i rygg och leder vid Rett syndrom, samt exempel på förebyggande och behandlande åtgärder.
Strategier för motorisk träning
Motivation och delaktighet
Effektiv träning kännetecknas av många repetitioner, tydlig struktur och att övningarna är lustfyllda. För att underlätta inlärning och utförande är det viktigt att personen har gott om tid att bearbeta instruktioner och har möjlighet att upprepa varje moment flera gånger för att stärka motoriskt minne. Trygghet är en förutsättning för god motorisk inlärning och det uppnås genom ett lugnt tempo, i känd miljö och med tillitsfulla relationer.
Balans mellan aktivitet och återhämtning
Det är viktigt med en bra balans mellan aktivitet och återhämtning för att minska risk för psykisk och fysisk överbelastning samt uppvarvande situationer. Aktivitetsnivån bör anpassas så att intensiva moment varvas med aktiviteter med låg stimuli. Varje människa har sitt sätt att varva ner. För att hjälpa en individ att återhämta sig och uppnå en känsla av lugn och ro i kroppen kan man försöka få till en lugn och djup andning och stimulera med känselintryck såsom tryck, beröring, vibration eller värme. Det kan uppnås med massage, varma fotbad eller att luta sig mot en vibrationskudde. Det kan även vara återhämtande att reducera intryck från omgivningen, till exempel att sitta i en koja, mysa med någon eller att lyssna på musik i hörlurar.
Sammanfattning
För att bibehålla och utveckla motoriska förmågor under hela livet är det viktigt att behandlingen är målinriktad och utförs kontinuerligt över tid. Detta inkluderar varierande fysiska aktiviteter, individuellt anpassande ortopediska hjälpmedel och tekniska lösningar samt regelbundna uppföljningar. En viktig utgångspunkt är att anpassa insatserna efter varje individs unika förutsättningar och behov samt utgå från personens intressen.
Det är vanligare med osteoporos vid Rett syndrom än för befolkningen generellt. Vid osteoporos har benvävnaden mer hålrum, är tunnare och svagare samt är lättare att bryta ner. Osteoporos ökar risken för frakturer och det är därför viktigt att följa upp vid Rett syndrom.
Osteoporos vid Rett syndrom
Personer med Rett syndrom har en ökad risk för osteoporos jämfört med befolkningen i allmänhet. Tillståndet kan uppkomma ganska tidigt hos personer med Rett syndrom och mutation i MECP2 genen, ibland redan i barnaåldern. Mutationer som är kopplade till svår form av Rett syndrom med avsaknad av gångförmåga och tidig skoliosdebut har också kopplats till ökad risk för osteoporos.
Kliniska riktlinjer för hantering av benhälsa vid Retts syndrom formulerades 2016 efter genomgång av tillgänglig litteratur och diskussion med experter. De ledde till 39 rekommendationer gällande bedömning och behandling. Artikeln och rekommendationerna finns att ladda hem här.
Faktorer som påverkar Osteoporos
Osteoporos beror på att balansen mellan uppbyggnad och nedbrytning av skelettet är rubbad vilket påverkas av olika faktorer. En av de viktigaste faktorerna som påverkar utveckling av benskörhet är kalcium och vitamin D-brist. Kalcium tillförs till kroppen med kost. Kalcium är viktigt, inte bara för skelettet, utan även för andra vävnader och organ i kroppen. För att kalcium ska kunna tas upp från maten till kroppen behövs vitamin D. Vitamin D bildas framför allt i huden med hjälp av solstrålarna men finns också i kosten. D-vitamin-brist leder till kalciumbrist vilket medför att kroppen börjar utnyttja kalcium som är lagrat i skelettet för att kunna fungera. Om detta pågår under längre tid blir skelettet alltmer urkalkat, tunnare och svagare.
En annan faktor som kan bidra till att osteoporos uppkommer är brist på fysisk aktivitet. Stillasittande eller stillaliggande personer riskerar att få en ganska snabb osteroporosutveckling även om nivån av vitamin D och kalcium är adekvat. Detta är viktigt att beakta vid Rett syndrom.
Nivåförändringar av könshormoner hos kvinnor under klimakteriet leder till att bentäthet minskar och efter menopausen är risken att utveckla benskörhet stor.
Benskörhet kan också uppkomma som biverkning vid längre medicinering med en del preparat, t.ex. kortisonpreparat, Valproatpreparat samt en del preventivmedel (depo medroxiprogesteronacetat – Depo Provera).
Ibland kan benskörhet vara ärftligt och vara vanligare i vissa släkter.
Vissa sjukdomar t.ex. ledgångsreumatism, glutenintolerans, sköldkörtelsjukdom ökar också risken för osteoporos. Det är viktigt att vara medveten om att dessa sjukdomar kan drabba personer med Rett syndrom liksom övriga befolkningen.
Mekanismen bakom utvecklingen av osteoporos hos personer med Rett syndrom är inte helt klarlagd. Förutom de ovanstående faktorerna finns det också djurstudier som visar att själva MECP2-proteinet påverkar benvävnadens utveckling och mineralisering och på så vis motverkar utveckling en av osteoporos. Brist på MECP2-protein skulle därför kunna öka risken för tidig osteoporosutveckling.
Typiska symtom
De första symtomen av kalcium- och D-vitaminbrist kan vara svårtydda skelett och muskelsmärtor. De blir ännu svårare att upptäcka/tolka hos personer med flerfunktionsnedsättning som har svårt att identifiera sina symtom och berätta om dem. Detta gäller även många personer med Rett syndrom. De första symtomen som omgivningen reagerar på hos den här gruppen är oftast frakturer som uppkommer vid lättare, eller ibland ingen, påfrestning (till exempel vid stretching).
Förutom att benbrott kan uppkomma i de långa skelettbenen (i armar eller ben) är ryggkotorna extra utsatta för osteoporos. Kompression av en kota är en av komplikationerna som kan uppkomma särskilt hos de personer som redan har extra belastning på ryggen på grund av t.ex. skolios. Et tecken på detta kan vara att kroppslängden plötsligt minskar med flera centimeter.
Diagnostik
Vitamin D och kalciumnivån i kroppen kan bedömas och följas med blodprover. Om riskfaktorer förekommer och/eller vid upprepade frakturer bör en speciell typ av röntgenundersökning, oftast av ländryggen och höfterna genomföras. Det är en mer avancerad och riktad undersökning som kallas för bentäthetsmätning eller DXA-mätning (Dual Energy X-ray absorptiometry). Först efter detta kan diagnosen osteoporos ställas.
Undersökningen används för att bedöma hur allvarlig osteoporosen är och för att avgöra vilken behandling som behövs och för uppföljning. Du kan läsa mer om undersökningen och hur den går till på 1177.
Förslag till åtgärder
Det är viktigt att kosten innehåller tillräcklig mycket kalcium och vitamin D. Kalcium finns framför allt i mejeriprodukter som mjölk, fil, yoghurt och ost men även i grönsaker samt spannmålsprodukter som pasta, bröd eller gryn.
Vitamin D finns också i maten, t.ex. i fisk eller fiskolja. Numera berikas även mjölk med vitamin D vid produktionen. Det är bra att rådgöra med en dietist om det finns behov av att komplettera kosten ytterligare. Vitamin D och kalcium tabletter kan förebygga att brist uppkommer. De kan också användas för bristbehandling, men vilka tabletter och vilken dos av vitamin D och kalcium som de ska innehålla bör bedömas av läkare.
En annan faktor som är mycket viktig och som kan motverka osteoporos utveckling är fysisk aktivitet. Aktiviteter som stimulerar att kroppen är i rörelse – bassängträning, ridning, eller även bara att gå (helst 2 timmar per dag) eller stå (minst 30 minuter), använda händerna så mycket som det går under vardagliga sysselsättningar – har positiv inverkan på kalciuminbyggnad i skelettet och minskar urkalkning. Daglig fysisk aktivitet är mycket viktig för att förebygga osteoporos och minska osteoporosens utvecklingstempo.
En del personer med uttalad osteoporos och upprepande frakturer behöver behandling med speciella preparat (bisfosfonater) vilket är skelettstärkande läkemedel som förbättrar bentäthet och kalkupptagning i benen.
Personer med Rett syndrom kan precis som alla andra drabbas av psykisk ohälsa och psykisk sjukdom. Psykisk ohälsa innebär per definition att personen i åtminstone någon utsträckning inte mår bra och är därför ett problem i sig. Det är också så att psykisk hälsa både kan påverkas av, och ha inverkan på viktiga delar av livet.
Brist på viktiga sociala relationer och meningsfulla aktiviteter kan leda till psykisk ohälsa och det är samtidigt vanligt att börja undvika viktiga sammanhang och personer när man själv inte mår bra. Svårigheter med sömn har en tydligt koppling till psykisk ohälsa både genom att sömnen är avgörande för att vi ska må bra både fysiskt och psykiskt samtidigt som psykisk ohälsa ofta har negativ inverkan på sömnen.
Det är lätt att psykisk ohälsa leder till minskad deltagande i sociala och andra hälsofrämjande aktiviteter. Vid misstanke om psykisk ohälsa och psykisk sjukdom är det viktigt att kontakta sjukvården för att få hjälp med en bedömning av specialister.
Forskningen kring psykisk ohälsa och Rett syndrom är begränsad men den forskning som finns pekar på att Rett syndrom innebär en förhöjd risk att utveckla psykisk ohälsa. I en amerikansk studie där man följt över 800 flickor och kvinnor med Rett syndrom uppvisade nästan alla deltagarna symtom på nedstämdhet och/eller ångestproblematik. Samtidigt var andra tecken på psykisk ohälsa såsom utåtagerande beteenden inte alls vanliga och dessutom mindre allvarliga.
När det gäller problem inom området depression och nedstämdhet visade intervjuer med närstående till 56 personer med Rett syndrom att lite drygt 75 procent av personerna med Rett syndrom hade nedstämdhet eller humörsvängningar. Hos nästan 15 procent var bekymren så påtagliga att de hade en fastslagen depressionsdiagnos. Förekomst av ångestsymtom har undersökts i flera studier och en litteraturöversikt med sammanlagt över 650 individer med Rett syndrom visade att över 70 procent åtminstone någon gång i livet haft någon form av ångestproblematik.
Psykisk ohälsa kan vara svår att upptäcka hos den som inte själv tydligt kan förmedla hur han eller hon mår. Det är viktigt att omgivningen är observant på förändringar hos personen. Beteendeförändringar som att personen blir mer lättirriterad och arg, får svårare att engagera sig i sådant som tidigare varit uppskattat eller att sömnmönstret ändras kan vara tecken på att personen inte mår bra.
Som nämnts ovan så behöver sjukvården göra en bedömning. Dels för att utesluta att den psykiska ohälsan orsakas av till exempelvis smärtsamma fysiska åkommor eller sjukdomar, dels för att få en bedömning av hur allvarlig personens psykiska hälsa är och om det är aktuellt med psykiatrisk diagnosticering. För bedömning och diagnosticering av psykiatriska tillstånd finns diagnoskriterier som anpassats för personer med intellektuell funktionsnedsättning, till exempel i diagnostiska manualen DM-ID-2.
Det finns en hel del som omgivningen kan göra för att främja psykisk hälsa och förebygga att psykisk ohälsa och psykisk sjukdom utvecklas. Grundläggande är att personen har sina medicinska och övriga basala behov, såsom sömn och näringsintag tillgodosedda. Det är också viktigt att personens vardag är begriplig, meningsfull och påverkbar. Personen behöver ges möjlighet att förstå vad som sägs och vad som ska hända. Aktiviteter behöver planeras utifrån vad personen tycker om och anpassas så att personen kan vara så delaktig som möjligt. I så stor utsträckning som möjligt behöver personen kunna påverka sitt liv genom att kommunicera både önskningar och behov. Mer information, förslag och inspiration finns under rubriker kommunikation, aktiviteter och kognitivt stöd.
Referenser
Edwards, G., Jones, C., Pearson, E., Royston, R., Oliver, C., Tarver, J., … & Waite, J. (2022). Prevalence of anxiety symptomatology and diagnosis in syndromic intellectual disability: A systematic review and meta-analysis. Neuroscience & Biobehavioral Reviews, 138, 104719.
Att äta är viktigt för livskvalitet och allmänt mående. Måltiden är också en stund för samvaro och för många ett sätt att umgås. Ät- och sväljsvårigheter är vanliga hos personer med Rett syndrom och det kan vara påfrestande för hela familjen och inte minst personen själv. Ät- och sväljsvårigheter påverkar också näringsintaget.
Ätsvårigheter förekommer i alla åldrar vid Rett syndrom men karaktären på svårigheter kan förändras genom åren. Att äta involverar muskler i munnen och svalget, munnens sensorik samt koordination med andning och sväljreflexer. Dessutom påverkas aptit och ätande av vårt allmänna mående. Det kan därför finnas en mängd olika orsaker till ätsvårigheter:
- Motoriska
- Sensoriska
- Medicinska
- Psykologiska
Motoriken i läpparna, tungan och svalget har stor betydelse för hur personen kan tugga och svälja. Låg muskeltonus (hypotonus) som är vanligt hos små barn med Rett syndrom kan ställa till svårigheter vid ätandet redan i tidig ålder. Vid låg muskelspänning är munnen ofta öppen i vila och läpparna inte slutna. Hypotonus kan påverka sugkraften vid amning och medföra att läpparna inte sluts när barnet tuggar och sväljer. Den första delen av ätandet när tuggan är i munnen sker viljemässigt medan sväljfasen, dvs när tuggan förts bakåt till svalget sker reflexmässigt.
Motoriska rörelser är beroende av sensoriska signaler (sinnesintryck) för att utvecklas och anpassas. En avvikande sensorik kan också innebära att exempelvis olika konsistenser eller smaker upplevs obehagliga.
Hälsotillstånd som gastroesofageal reflux, förstoppning, hjärtsjukdom eller öroninfektioner är kopplade till ätsvårigheter. En del mediciner kan också påverka aptiten och ätandet.
Oberoende av orsaken till ätsvårigheter kan negativa upplevelser kopplat till mat, dryck och ätande leda till att personen undviker att äta eller utvecklar aversion mot viss mat, konsistens eller smak. Ätsvårigheter upplevs ofta mycket stressande för föräldrar och det är inte ovanligt att ätande leder till kamp och konflikt.
På Mun-H-Centers hemsida finns mer att läsa om ätande och ätsvårigheter.
Munmotorik och ätsvårigheter vid Rett syndrom
Forskning visar att ätsvårigheter är vanligt vid Rett syndrom och kan förekomma även när närstående rapporterar att ätandet fungerar bra. Ofrivilliga tungrörelser har visat sig vara vanligt (ca 75%) liksom begränsade tungrörelser, exempelvis att tuggan inte förs till tänderna med hjälp av tungan. Ätandet behöver dessutom samordnas med andningen. Andningsstörningarna som är vanligt förekommande vid diagnosen påverkar därför också ätandet och äthastigheten. De motoriska svårigheterna innebär att tuggan eller dryck ibland sätts i halsen. Cirka 5-8% av personerna med Rett syndrom har stora sväljsvårigheter med hög risk att dra ner mat i luftvägarna men för de allra flesta med Rett syndrom fungerar sväljfasen relativt bra liksom hostreflexen som är en del av att skydda luftvägarna.
Mellan 60-80% av personerna med Rett syndrom gnisslar tänderna (bruxism) och särskilt de som gnisslar tänderna dagtid behöver därför oftare tandvård. En åtgärd är bettskena för att skydda tänderna. I ett par fallbeskrivningar rapporteras positiva resultat av botox och i en annan av akupunktur i kombination med bettskena.
Behandling och åtgärder
Vid tecken på ätsvårigheter är det viktigt att ett tvärprofessionellt team utreder orsaker och föreslår behandling och åtgärder. Sådana tecken kan exempelvis vara att måltiden tar lång tid (över 30 minuter), upplevs jobbig/stressig, att barn inte går upp i vikt, viktnedgång hos både barn och vuxna, hosta eller rosslighet i samband med måltid eller återkommande lunginflammationer. Vid en utredning är ofta flera professioner inkopplade, bl.a. logoped, dietist och läkare men även andra professioner kan ingå och i många regioner finns särskilda team med specialistkunskap om ätsvårigheter.
Utifrån utredningen kan teamet ge rekommendationer om hur personen bäst ska kunna äta säkert och så lustfyllt som möjligt. En viktig del av åtgärderna är att förebygga att måltider och ätande blir negativa upplevelser vilket kan befästa eller öka ätsvårigheterna. Det finns också träningsprogram för att stimulera eller träna sensorik, motorik och ätförmåga som teamet kan rekommendera om de är lämpliga för den specifika individen. Anpassade bestick, muggar och tallrikar kan underlätta för personen att äta själv eller att vara mer delaktig i att föra maten till munnen. Vid misstanke om aspiration kan fortsatt utredning med exempelvis videoröntgen vara aktuell. Vid stora ätsvårigheter när det är svårt att få i sig tillräckligt med näring kan det bli aktuellt med gastrostomi, d.v.s. att näring tillförs i en sond via en knapp på magen. Beroende på personens lust att äta, ätsvårigheter och risk för aspiration (att mat dras ner i luftstrupen och lungorna) kan stora delar, smakportioner eller ingen näring alls via munnen vara aktuellt.
Här är några generella råd gällande ätande och måltider som är bra för många med ätsvårigheter (men inte alla).
- Fokusera på det som är positivt med måltiden. Visa när du njuter av din mat och prata positivt om ätande och måltid. Undvik att prata om personens ätsvårigheter vid matbordet när personen är närvarande.
- Rutiner underlättar för personen att veta vad som förväntas.
- Se till att allt är klart när personen sätter sig vid bordet för att undvika väntan om det upplevs jobbigt.
- Håll måltiderna korta (max 20/30 minuter) om inte personen själv njuter av att sitta till bords.
- Servera små portioner och ta om hellre än att lägga upp en stor portion. Det kan upplevas mer överkomligt för både personen själv och den som eventuellt assisterar.
- Uppmuntra personen att smaka på saker men tvinga inte.
- Se till att personen sitter så stabilt som möjligt och att huvudet inte är bakåtlutat eftersom det ökar risken att sätta i halsen.
- För barn: undvik i möjligaste mån att använda pipmugg eftersom det befäster ett omoget tungmönster vilket hindrar ett mer effektivt och aktivt sätt att använda tungan och läpparna. Logoped kan vara behjälplig i detta.
- För personer i omgivningen kan det vara frestande att komma med goda råd. Bidra hellre med praktiskt stöd. Fråga vad du kan göra för att avlasta, exempelvis diska upp efter maten så att familjen kan göra något tillsammans den stunden istället eller få tid för återhämtning.
Läs mer
Sömnsvårigheter är mycket vanliga hos personer med Rett syndrom och avvikande sömnmönster är listat som ett stödjande kriterium vid diagnosticering. Exempel på sömnproblem är insomningssvårigheter, nattliga uppvaknanden, överdrivet stort sömnbehov även under dagtid samt alldeles för lite eller mycket total sömntid under dygnet.
Beroende på vad som inkluderas i studier av sömnsvårigheter så varierar rapporter om förekomst. I en studie från Australien där forskarna inhämtat enkätsvar i omgångar mellan åren 2000 och 2011 fann man att generella sömnproblem förekom hos ca 90 procent hos barn och ungdomar medan genomsnittlig förekomst var något lägre hos vuxna. Hos barn upp till 7 år brukade nästan 80 procent vakna minst någon gång per vecka och skratta utan synbar anldening medan siffran sjunkit till cirka 60 procent bland de över 18 år. Förekomst av nattliga uppvaknanden med skrikande var knappt 50 procent hos de små barnen och ca 40 procent bland vuxna.
Sömnen är viktg för alla och sömnsvårigheter har ofta inverkan på hur vi människor fungerar under dagtid. Sömnbrist resulterar såklart i trötthet och i forskning är det vanligt att man ser samband mellan sömnsvårigheter och mer eller mindre allvarliga problematiska beteenden under dagtid. Sömnsvårigheter drabbar såklart personen själv men även ofta hela familjen och det är viktigt att man hittar vägar att förbättra sömnen.
I första hand brukar man rekommendera icke-medicinska interventioner och man pratar då ofta om att förbättra sömnhygienen (sleep hygiene på engelska). Sömnhygien kan sägas vara ett paraplybegrepp som innefattar faktorer som påverkar en persons sömn. Dessa faktorer kan delas in i fyra olika kategorier.
- Miljömässiga faktorer, till exempel temperatur i sovrummet, belysning och ljudnivå.
- Planering, till exempel regelbundna tider och bra balans mellan sömn och vakentid.
- Sömnrelaterade vanor, till exempel kvällsrutiner som signalerar och förbereder för sänggående.
- Fysiologiska faktoerer, till exempel aktivitetsnivå under dag och kväll samt planering av mat och måltider.
Att se över och justera sömnhygien är en bra början för att hjälpa personen att sova bättre. Det är dock inte ovanligt att sömnhygieniska strategier behöver kompletteras med medicinsk behandling i samråd med läkare. Det kan också vara bra att ta kontakt med vården för att utesluta eventuella medicinska eller kroppsliga orsaker till sömnproblemen.
För att kunna planera insatser och genomföra effektiva uppföljningar är det viktigt att få en tydlig bild av personens sömnvanor. Sömndagbok har visat sig vara ett bra sätt att samla information som sedan används för att planera insatser. Sömndagboken ska fyllas i tills man ser ett stabilt mönster och forskning har visat att mönster brukar kunna framträda efter att registrering under fem vardagsnätter i rad. Oavsett om man vill prova att förändra sömnhygien på egen hand eller söka stöd hos till exempel habiliteringen så är det bra att börja registrera i en sömndagbok. Ta med den ifyllda dagboken till läkarbesöket eller använd den som utgångspunkt för att prova strategier på egen hand.
Här finns ett exempel på en sömndagbok som kan skrivas ut och användas direkt eller anpassas efter era behov.
Referenser
Heussler, H. S. (2016). Management of sleep disorders in neurodevelopmental disorders and genetic syndromes.
Current Opinion in Psychiatry,
29(2), 138-143.
Jan, J. E., Owens, J. A., Weiss, M. D., Johnson, K. P., Wasdell, M. B., Freeman, R. D., & Ipsiroglu, O. S. (2008).
Sleep hygiene for children with neurodevelopmental disabilities. Pediatrics, 122(6), 1343–1350. https://doi.org/10.1542/peds.2007-3308
Referenser rörande sömndagbok finns på hemsidan dit länken leder.

Samhällsstöd
För att underlätta livet med en sällsynt diagnos finns det stöd att få, men informationen kan vara svår att hitta. Här samlar vi information som vi tror är behjälplig för personer med Rett syndrom och deras familjer. Lagen om stöd och service till vissa funktionshindrade (LSS) ligger till grund för samhällets stöd och olika typer av stöd kan vara aktuella under olika faser i livet.

Forskning
Ett allt större antal studier pågår för att öka kunskapen om Rett syndrom. Många studier handlar om att försöka koppla ihop genetik med symtom eller att beskriva hur vanligt det är att olika symtom förekommer inom gruppen. Allt fler artiklar handlar om behandling och andra insatser för att lindra symtom och att stötta utveckling. I dessa studier prövas både medicinsk behandling, träning av olika färdigheter och pedagogiska insatser. Forskning för att bota eller hindra förloppet av diagnosen pågår också. Vi försöker hålla oss uppdaterade och länkar både till svensk och internationell forskning. För vissa forskningsartiklar kan det krävas betalning
Vid frågor om forskningsartiklar är ni välkomna att kontakta oss.
Mer information
Mer om Rett syndrom, sällsynta diagnoser, funktionsnedsättning och att vara anhörig hittar du på hemsidorna som är listade nedan.
Sidan uppdaterades senast: 25 maj, 2026